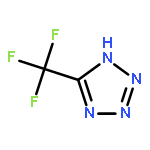
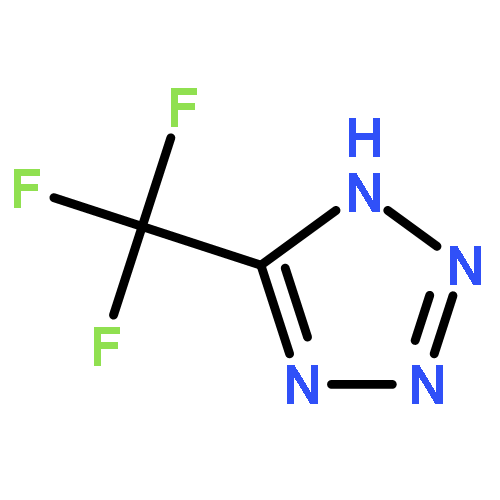
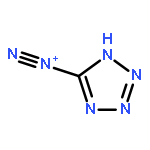
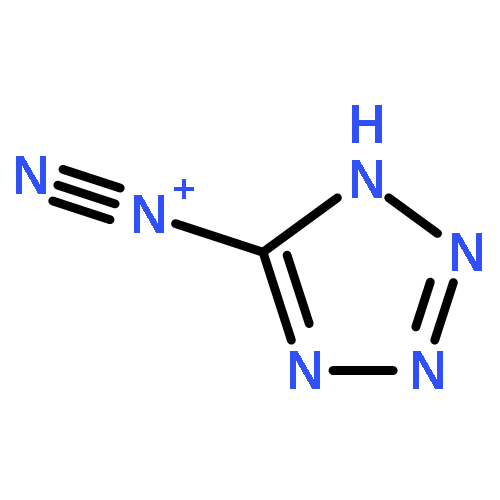
Co-reporter:Sheng-En Zhang, Feng Cheng, Xiang He, and Zhao-Xu Chen
The Journal of Physical Chemistry C October 26, 2017 Volume 121(Issue 42) pp:23463-23463
Publication Date(Web):October 2, 2017
DOI:10.1021/acs.jpcc.7b07461
Reactive molecular dynamics simulations were performed to explore the structural evolution of a ZnO nanocluster and (0001) and (101̅0) surfaces under a H2 atmosphere at different temperatures. The mechanisms of H2 dissociation and water formation were analyzed. Our simulations reveal that there are two pathways for H2 dissociation and three routes for water formation on the surfaces. The nanocluster is more active for H2 dissociation and water formation than the two surfaces. The gas–solid interactions lead to outward displacement of the substrate O atoms. While the O-terminated surface of the (0001) facet is active for H2 dissociation and water formation, the Zn-terminated one is inactive for the dissociation. Unlike the (0001) surface which is more easily reduced, the (101̅0) surface is readily hydroxylated. Water formation and desorption results in surface oxygen depletion and Zn aggregation which lead to surface metallization, in accordance with the experimental observations. Our simulations show that Zn sites are not active for H2 dissociation. By fitting the obtained rate constants at different temperatures, we estimated the activation energy of the H2 dissociation over ZnO cluster to be 4.06 kcal/mol, in very good agreement with the experimental result of 5.0 kcal/mol.
Co-reporter:Xiang He;Guo-Jun Kang
The Journal of Physical Chemistry C July 16, 2009 Volume 113(Issue 28) pp:12325-12330
Publication Date(Web):Publication Date (Web): June 17, 2009
DOI:10.1021/jp9006729
Selective hydrogenation of the C═O group of acrolein produces the chemically important intermediate propenol. Addition of the second metal indium to gold catalyst notably improves the selectivity to the desired ally alcohol. The role of indium was previously ascribed to block active sites on faces for the C═C hydrogenation by formation of an inactive indium phase. We employed density functional slab models to investigate acrolein adsorption and hydrogenation on pure and indium-decorated Au(110) surfaces. It is shown that, due to a stronger In−Au interaction, indium favors deposition on faces over corners or edges. Acrolein interacts more strongly with the decorated gold surfaces via O−In interaction than with the clean gold surface. Comparison reveals that C═C (C═O) hydrogenation prefers sites at edges (faces). The site preference of indium and the strong O−In interaction greatly increases (decreases) the concentration of acrolein at faces (edges), which improves the selectivity to propenol significantly.
Co-reporter:Xiang He, Feng Cheng, Zhao-Xu Chen
Applied Surface Science 2017 Volume 425(Volume 425) pp:
Publication Date(Web):15 December 2017
DOI:10.1016/j.apsusc.2017.06.256
•(CH3)2S adsorption on Au(111) was calculated with density functional method.•The effect of electric field on (CH3)2S adsorption was examined.•The mechanism of the substrate work function reduction is theoretically explored.•The work function of Au gate sensitively correlates the content of (CH3)2S.The interface interaction between the dimethyl sulfide (DMS) molecule and the gold substrate under external electric fields is investigated by density functional theory method. The polarized DMS adsorbate reduces the work function of the gold substrate while the induced substrate dipole upon the adsorption slightly increases the work function. The DMS layer partially shields the Au(111) substrate from the electric fields and the vacuum level of DMS/Au(111) shifts less than of Au(111) in consequence. Under electric fields pointing outward from the Au(111) surface, both the reduction of work function and the adsorption of DMS molecule are enhanced on the surface. We also suggest the possible application of the field-effect transistor (FET) sensor with gold gate for detecting DMS molecule by utilizing the reduction of substrate work function upon adsorption. The effects of coverage and electric field on the theoretical sensitivity of the sensor are also discussed.Download high-res image (132KB)Download full-size image
Co-reporter:Feng Cheng and Zhao-Xu Chen
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 5) pp:3936-3943
Publication Date(Web):22 Dec 2015
DOI:10.1039/C5CP05020K
Pd/ZnO is a promising catalyst studied for methanol steam reforming (MSR) and the 1:1 PdZn alloy is demonstrated to be the active component. It is believed that MSR starts from methanol dehydrogenation to methoxy. Previous studies of methanol dehydrogenation on the ideal PdZn(111) surface show that methanol adsorbs weakly on the PdZn(111) surface and it is hard for methanol to transform into methoxy because of the high dehydrogenation barrier, indicating that the catalyst model is not appropriate for investigating the first step of MSR. Using the model derived from our recent kinetic Monte Carlo simulations, we examined the process CH3OH → CH3O → CH2O → CHO → CO. Compared with the ideal model, methanol adsorbs much more strongly and the barrier from CH3OH → CH3O is much lower on the kMC model. On the other hand, the C–H bond breaking of CH3O, CH2O and CHO becomes harder. We show that co-adsorbed water is important for refreshing the active sites. The present study shows that the first MSR step most likely takes place on three-fold hollow sites formed by Zn atoms, and the inhomogeneity of the PdZn alloy may exert significant influences on reactions.
Co-reporter:Yan-Hua Lei, Zhao-Xu Chen
Applied Surface Science 2016 Volume 361() pp:107-113
Publication Date(Web):15 January 2016
DOI:10.1016/j.apsusc.2015.11.057
Highlights
- •
Under normal experimental conditions perfect surface of MoO3(0 1 0) is favorable.
- •
Line defects along asymmetric oxygen direction in lean oxygen condition are favored.
- •
Vacancy replenishing occurs on vacancies formed by terminal and asymmetrical oxygen.
Co-reporter:Yuping Cai, Sheng’en Zhang, Yunsheng Xue, Juli Jiang, and Zhao-Xu Chen
The Journal of Organic Chemistry 2016 Volume 81(Issue 5) pp:1806-1812
Publication Date(Web):January 21, 2016
DOI:10.1021/acs.joc.5b02503
Cu-catalyzed cross-dehydrogenative coupling (CDC) reaction of thiazoles with THF has been studied with the density functional theory method and kinetic Monte Carlo (kMC) simulations. Our results show that the previously proposed concerted metalation–deprotonation mechanism is unfavorable. On the basis of the DFT calculation and kMC simulation results, a new mechanism is proposed. In the favorable mechanism, the Cu(II) catalyst first combines with the thiazoles, forming an organocopper species that then binds to the THF radical. The rate-limiting step, C–C bond formation, is realized through an intramolecular structural rearrangement. The Cu catalyst works as a matchmaker to render the C–C bond formation. Kinetic Monte Carlo simulations demonstrate that one should be careful with the conclusions drawn simply from the calculated barriers.
Co-reporter:Yan-Hua Lei
Science China Chemistry 2015 Volume 58( Issue 4) pp:593-600
Publication Date(Web):2015 April
DOI:10.1007/s11426-015-5341-x
The role of bismuth in the selective oxidation of propene has long been debated. We performed density functional calculations to study the dehydrogenation reaction of propene on Bi2O3 surfaces. Our calculated thermodynamic data reveal that the first dehydrogenation of propene on the most stable (010) surface and the (100) surface are difficult. Our calculations indicate that the barrier of the first hydrogen abstraction on the high Miller index surface (211) is much lower than those on the (100) and (010) surfaces, and is close to the experimental one. Further dehydrogenation is shown to be difficult and production of 1,5-hexadiene through dimerization of allyl is likely, in agreement with the experimental observations.
Co-reporter:Feng Cheng ;Dr. Zhao-Xu Chen
ChemCatChem 2015 Volume 7( Issue 13) pp:1926-1930
Publication Date(Web):
DOI:10.1002/cctc.201500366
Abstract
Two-monolayer-Zn covered Pd(1 1 1) annealed at (low) 500 K exhibits nice CO2 selectivity for CH3OH+H2O to CO2+H2, whereas CO is yielded exclusively when annealed at (high) 650 K. To unravel the reason behind the phenomenon, kinetic Monte Carlo (KMC) simulations were used to study the alloying process. It shows the low temperature annealing produces a multilayer 1:1 PdZn alloy, whereas the high temperature operation results in a Zn-lean multilayer alloy, not the previously assumed monolayer PdZn alloy on Pd. The geometry and electronic structures of the models derived from KMC simulation agrees with the relevant experiments. The low temperature sample is more active than the high temperature one for H2O dissociation, in line with the assumption that H2O dissociation controls CO2 selectivity. It is revealed that triple Zn ensembles which form three-fold hollow sites account for the activity for H2O dissociation.
Co-reporter:Zengyang Xie, Yuping Cai, Hongwen Hu, Chen Lin, Juli Jiang, Zhaoxu Chen, Leyong Wang, and Yi Pan
Organic Letters 2013 Volume 15(Issue 17) pp:4600-4603
Publication Date(Web):August 22, 2013
DOI:10.1021/ol4022113
Copper-catalyzed cross-dehydrogenative coupling (CDC) reactions of (benzo)thiazoles with cyclic ethers were developed under mild conditions. In particular, the formation of C–C bonds via the CDC reactions between non-benzo-fused azoles and ethers are reported for the first time. In addition, the acetals, known as the masked 2-thiazolecarboxaldehydes, could be successfully obtained by this CDC reaction. The preliminary mechanism and supportive DFT calculations are discussed as well.
Co-reporter:Xiufang Ma;Yanhua Lei;Zhaoxu Chen
Chinese Journal of Chemistry 2013 Volume 31( Issue 3) pp:421-429
Publication Date(Web):
DOI:10.1002/cjoc.201201107
Abstract
Functionalization of the inert CH bonds of unsaturated molecules by transition metal complex is an important means to form new CC bonds. The functionalization is usually initiated by the ligand dissociation of a complex. In this paper we employ both ab initio and density functional methods to explore the influence of central metals, conformation, solvent and protonation on the ligand dissociation of the (hfac-O,O)2M(L)(py) complexes [M=Rh(III) or Ir(III), hfac-O,O=k2-O,O-1,1,1,5,5,5-hexafluoroacetylacetonato, L=CH3, CH3CO2, (CH3CO)2CH, CH3O or OH, py=pyridine]. We demonstrate that ligand pyridine dissociates more easily than the "L" ligands under study in aprotic solvent and gas phase and the dissociation of pyridine is more facile in the trans-conformation than in the cis-isomer. These phenomena are rationalized based on electronic structure and molecular orbital interactions. We show that solvation only slightly stabilizes the complexes and does not change the ligand dissociation ordering. In particular, we show that pyridine is no longer the labile ligand in protic media. Instead, the oxygen-containing ligands (apart from those like hfac that form a cyclic structure with the central metal) that coordinate to the central metal via oxygen atom become the labile ones. Finally our calculations indicate that hfac is a stable ligand, even in protic media.
Co-reporter:Tao Tao, Yan-Hua Lei, Yu-Xin Peng, Ying Wang, Wei Huang, Zhao-Xu Chen, and Xiao-Zeng You
Crystal Growth & Design 2012 Volume 12(Issue 9) pp:4580-4587
Publication Date(Web):July 11, 2012
DOI:10.1021/cg300776y
Metal-directed assembly of naphthalene-1,4,5,8-tetracarboxylic acid (NTA) with different transition-metal salts in the presence of ammonia results in a series of one-dimensional metal–naphthalenediimidato (M–NDI) coordination polymers with the formulas of {[Ag(NDI)](NH4)}n (P1), [Zn(NDI)(NH3)2]n (P2), [Cd(NDI)(NH3)2]n (P3), [Co(NDI)(NH3)2]n (P4) and [Ni(NDI)(NH3)2]n (P5), respectively. It is worthwhile to mention that the 1D straight-line NDI–Ag(I) coordination polymer P1 is formed stepwise from a dinuclear NDI–Ag(I) intermediate [Ag2(NDI)(NH3)2] (2AgNDI), where ammonia serves as a stabilizing reagent of Ag(I) ion and a weak base to remove the protons of NDIH2 simultaneously. Furthermore, P1 exhibits semiconducting properties in the solid state which may originate from its all-parallel-aligned packing structure (AAAA) which is different from the common ABAB packing mode for P2–P5 and 2AgNDI. In addition, theoretic computational studies as well as X-ray photoelectron spectrometer spectra on P1 and 2AgNDI have also been carried out.
Co-reporter:Guo-Jun Kang;Jing Ma
Catalysis Letters 2012 Volume 142( Issue 2) pp:287-293
Publication Date(Web):2012 February
DOI:10.1007/s10562-011-0755-3
Unsaturated alcohols, usually produced from selective hydrogenation of unsaturated aldehydes, are important fine chemical intermediates used to synthesize pharmaceuticals and flavoring materials. Acrolein, the smallest member in α, β-unsaturated aldehydes, is the model system for studying selective hydrogenation of α, β-unsaturated aldehydes. So far most theoretical work is about adsorption and reactions of acrolein and its related species on surfaces. In the present paper we systematically studied the geometries, electronic structures, stability and transformation of various species derived from stepwise hydrogenation of acrolein in the gas phases. We identified the most stable intermediates for each system and determined the energy barrier for intermolecular conversion between isomers for various species with different content of hydrogen. All these results are valuable and informative for understanding the surface chemistry of hydrogenation of α, β-unsaturated aldehydes.
Co-reporter:Yan-Hua Lei and Zhao-Xu Chen
The Journal of Physical Chemistry C 2012 Volume 116(Issue 49) pp:25757-25764
Publication Date(Web):November 19, 2012
DOI:10.1021/jp304122n
MoO3 is an important catalytic material, and there exist controversial viewpoints about its surface structure, oxygen vacancy, and hydrogen adsorption, which are crucial for rationalizing the catalytic properties and reaction mechanism. To clarify these disputes, we adopted the density functional plus U (DFT+U) method to investigate properties of MoO3 bulk and surfaces and examined atomic hydrogen adsorption. Analyses reveal that the vibration peak at 820 cm–1 previously assigned to the vibration of asymmetrical oxygen is due to the vibration of symmetrical oxygen. On the other hand, the previously unassigned weak peaks at 899 and 723 cm–1 are caused by the asymmetrical oxygen stretching. Single hydrogen atom adsorbs favorably at asymmetric oxygen, while the terminal oxygen becomes the favorable position for accommodating two hydrogen atoms. The H atoms occupy preferentially asymmetrical oxygen at low coverage, whereas at high coverage they favorably reside on the terminal one. Our calculations indicate that different from the previous viewpoint, water binds to the terminal oxygen defective site relatively strongly. Furthermore, the controversial viewpoints about the stability ordering of oxygen vacancy under oxidation and reduction conditions is discussed on the basis of the formation energy of oxygen vacancy and water desorption energy on defect sites.
Co-reporter:Yeqian Shen, Yucheng Huang, Sujuan Zheng, Xuefeng Guo, Zhao-Xu Chen, Luming Peng, and Weiping Ding
Inorganic Chemistry 2011 Volume 50(Issue 13) pp:6189-6194
Publication Date(Web):May 31, 2011
DOI:10.1021/ic200459t
CeVO4 nanocrystals doped by heteroions were prepared via a hydrothermal method without the presence of surfactants or templates. Transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), X-ray diffraction (XRD), solid state 51V NMR, and inductively coupled plasma (ICP) were used to characterize the morphology, structure, and compositions of the materials. X-ray photoelectron spectroscopy (XPS) results confirmed that there is a valence change from Ce3+ to Ce4+ for a fraction of cerium atoms whereas the vanadium atoms remain in the pentavalent state V5+ upon the substitution of Ca2+ into CeVO4. Raman spectroscopy was used to monitor the effects of the doping ion on the CeVO4 lattice contraction and distortion. The appearance of the shifted and broadened Raman peaks for the doped CeVO4 was interpreted by theoretical calculations performed with Vienna ab initio simulation package. The redox properties and photocatalytic activities of the obtained nanocrystals were also investigated and discussed in detail.
Co-reporter:Xiang He, Yucheng Huang and Zhao-Xu Chen
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 1) pp:107-109
Publication Date(Web):16 Nov 2010
DOI:10.1039/C0CP01344G
Catalytic performances of alloy and surface alloy are sensitive to the surface structures and composition. In this paper we present an overall survey of the surface structure of Pd(111) covered with different amount of Zn using Monte Carlo simulations. We demonstrate that the composition of PdZn surface alloy is Zn coverage dependent: the surface concentration of Zn increases with the increase of the deposited Zn. At one or multi-layer of zinc deposited Pd(111), a multilayer 1:1 PdZn surface alloy will be formed. Surface alloy islands dominated by palladium are formed at submonolayer Zn coverage. At very low zinc coverage, small palladium ensembles of 3 to 5 Pd atoms exist preferentially on the Pd(111) surface. Our simulated results which are consistent with the pertinent experiments indicate that the unusual high-temperature desorption peak of formaldehyde observed experimentally has likely originated from the small surface ensembles induced by deposited Zn.
Co-reporter:Zhongqiang Liu, Zhao-Xu Chen, Biaobing Jin, Xianxi Zhang
Vibrational Spectroscopy 2011 Volume 56(Issue 2) pp:210-218
Publication Date(Web):18 July 2011
DOI:10.1016/j.vibspec.2011.02.010
Electronic structure calculations and spectroscopic assignments for metallophthalocyanines NiPc, PdPc and PtPc are performed on optimized geometries at B3LYP/LANL2DZ level. The order of the sizes of the central hole is computed to be PdPc > PtPc > NiPc, with the hole size of PdPc close to that of PtPc. The Mulliken charges of the central M vary in the order of PtPc > NiPc > PdPc, and the HOMO–LUMO gaps are in the order of NiPc < PdPc < PtPc, in agreement with the experimental result. The simulated IR spectra for the three derivatives are compared with the experimental absorption spectra, and very good consistency has been obtained. The simulated medium intensity bands associated with the metal–ligand vibrations which appear as singlet bands at 880, 877 and 883 cm−1, respectively, exhibit the order of PtPc > NiPc > PdPc, which is the same order as experiment. Furthermore, the metal–ligand vibrational bands for Raman spectra shift in the order NiPc > PtPc > PdPc. The strongest Raman lines predicted at 1562, 1532 and 1534 cm−1 for NiPc, PdPc and PtPc are very sensitive to the metal ion.
Co-reporter:Yucheng Huang
The Journal of Physical Chemistry C 2011 Volume 115(Issue 38) pp:18752-18760
Publication Date(Web):August 18, 2011
DOI:10.1021/jp206093e
Recently, it was proposed that the different behavior of water on PdZn multilayer and monolayer is responsible for the different selectivity toward CO2 in methanol steam reforming process on the Zn deposited Pd surface annealed at different temperatures. [Angew. Chem.2010,49 (18), 3224–3227.] To explore this interesting and important phenomenon, we investigate water adsorption and dissociation in various aggregation forms on PdZn multi- and monolayer model surface alloys of both flat and stepped surfaces, using density functional theory and slab models. Our calculations show that the water is more stable on the multilayer. Surface defects and aggregation favor H2O dissociation. Contrary to the point of view that PdZn monolayer cannot activate H2O whereas multilayer can, our first-principles results clearly demonstrate that the multilayer is less active for water dissociation than the monolayer. This discrepancy calls for further studies on this system both experimentally and theoretically.
Co-reporter:Guo-Jun Kang;Zhe Li
Catalysis Letters 2011 Volume 141( Issue 7) pp:996-1003
Publication Date(Web):2011 July
DOI:10.1007/s10562-011-0569-3
We studied acrolein (AC) adsorption on gold clusters Aun (n = 1–5) using density functional theory. It is demonstrated that conjugation effect reduces the adsorbate–substrate interaction through π-(C=C), π-(C=O) and di-σ-(C=O) modes whereas it facilitates the di-σ-(C=C) and the σ-O configurations. Analysis reveals that in π-(C=C) and π-(C=O) modes acrolein uses the HOMO-1 orbital to interact with the clusters while in σ-O mode the HOMO of AC plays the role. For di-σ-(C=C), di-σ-(C, O) and di-(C=O), the HOMO orbital of the cluster donates electrons to acrolein. Acrolein adsorption through the C=C bond is more favorable than that via the C=O group, which explains why the yields of C=C hydrogenation is higher than that of C=O reduction.
Co-reporter:Yucheng Huang and Zhao-Xu Chen
Langmuir 2010 Volume 26(Issue 13) pp:10796-10802
Publication Date(Web):April 26, 2010
DOI:10.1021/la100619q
Methanol dehydrogenation on Pd(111) and various Pd−Zn surface alloy films supported on Pd(111) have been investigated using density functional method in combination with periodic slab models. Calculations show that compared to Pd(111) the interaction between CH3O and the films is enhanced, whereas that for CH2O and CHO is weakened. Zn in top layer facilitates the CH3O stability. At variance, the subsurface Zn reduces the interaction of CH2O and CHO with the substrate significantly. Addition of Zn promotes the O−H breaking of CH3OH and the dehydrogenation of CHO but hinders the dehydrogenation of CH3O and CH2O. Comparison shows that the third-layer Zn atoms have essentially no effect on the reactions. Our calculations demonstrate that the experimentally observed 360 K desorption peak cannot be originated from CH2O adsorbed at flat Pd−Zn alloy surfaces, and it is very likely that CH2O combines preferentially with some species before decomposing into CHO during methanol steam reforming if CH2O is an intermediate. Finally, we show that the newly proposed relationship between the energy of the initial states and transition states exhibits better correlation than the classical BEP relation.
Co-reporter:Zhongqiang Liu, Zhao-Xu Chen, Weiping Ding, Guo-Jun Kang, Zhe Li
Journal of Molecular Structure: THEOCHEM 2010 Volume 948(1–3) pp:99-101
Publication Date(Web):30 May 2010
DOI:10.1016/j.theochem.2010.02.028
Incorporation of P into ZSM-5 framework greatly improves the catalytic properties of the molecular sieve. To understand the feasibility of existence of pentacoordinated phosphorus species in ZSM-5, we designed a model reaction and studied it using density functional theory at B3LYP/6-31G∗ level to evaluate its thermodynamics. The result demonstrates that the formation of pentacoordinated phosphorus species is thermodynamically favorable at high temperatures by hydrothermal treatment of the ZSM-5 modified with phosphorus acid.
Co-reporter:Qian-Lin Tang, Zhao-Xu Chen, Xiang He
Surface Science 2009 Volume 603(Issue 13) pp:2138-2144
Publication Date(Web):1 July 2009
DOI:10.1016/j.susc.2009.04.011
The water gas shift (WGS) reaction is an important reaction system and has wide applications in several processes. However, the mechanism of the reaction is still in dispute. In this paper we have investigated the reaction mechanism on the model Cu(1 1 1) system using the density functional method and slab models. We have characterized the kinetics and the thermodynamics of the four reaction pathways containing 24 elementary steps and computed the reaction potential energy surfaces. Calculations show that the formate (HCOO) intermediate mechanism (CO + OH → HCOO → CO2 + H) and the associative mechanism (CO + OH → CO2 + H) are kinetically unlikely because of the high formation barrier. On the other hand, the carboxyl (HOCO) intermediate mechanism (CO + OH → HOCO → CO2 + H) and the redox mechanism (CO + O → CO2) are demonstrated to be feasible. Our calculations also indicate that surface oxygen atoms can reduce the barriers of both water dissociation and HOCO decomposition significantly. The calculated potential energy surfaces show that the water dissociation which produces OH groups is the rate-determining step at the initial stage of the reaction or in the absence of surface oxygen atoms. With the development of the reaction or in the presence of oxygen atoms on the surface, CO + OH → HOCO and CO + O → CO2 become the rate-limiting step for the carboxyl and redox mechanisms, respectively.
Co-reporter:Qian-Lin Tang, Zhao-Xu Chen
Surface Science 2007 Volume 601(Issue 4) pp:954-964
Publication Date(Web):15 February 2007
DOI:10.1016/j.susc.2006.11.036
Density functional and periodic slab model calculations are performed to study adsorption of water on various Cu surfaces, focusing on monomers and dimers at the planar Cu surfaces and monomers at stepped ones. Single water molecules tend to weakly bind to atop positions with the molecular plane basically parallel to the substrate surface on the planar surfaces or the step plane on the stepped surfaces with negligible structural deformation of water. The experimental adsorption energies of water on the (1 1 1) and (1 0 0) surfaces are about twice as large as the theoretical values of monomerically adsorbed water. This phenomenon is demonstrated to be due to formation of water clusters and/or existence of surface defects. It is revealed that the most favorable hexagonal ring superstructure on Cu(1 1 0) is a four-layer structure, not the commonly accepted bi-layer configuration. We found that the adsorption energy of monomeric water correlates linearly with following quantities, respectively: the bond length and the stretching frequency of the Cu–O bond, the coordination number of the surface Cu atom, the surface work function of the clean surface and the 1b1 MO energy shift with respect to the value in the gas phase.
Co-reporter:Yilin Cao, Zhao-Xu Chen
Surface Science 2006 Volume 600(Issue 19) pp:4572-4583
Publication Date(Web):1 October 2006
DOI:10.1016/j.susc.2006.07.028
To provide information about the chemistry of water on Pd surfaces, we performed density functional slab model studies on water adsorption and decomposition at Pd(1 1 1) surface. We located transition states of a series of elementary steps and calculated activation energies and rate constants with and without quantum tunneling effect included. Water was found to weakly bind to the Pd surface. Co-adsorbed species OH and O that are derivable from H2O stabilize the adsorbed water molecules via formation of hydrogen bonds. On the clean surface, the favorable sites are top and bridge for H2O and OH, respectively. Calculated kinetic parameters indicate that dehydrogenation of water is unlikely on the clean regular Pd(1 1 1) surface. The barrier for the hydrogen abstraction of H2O at the OH covered surface is approximately 0.2–0.3 eV higher than the value at the clean surface. Similar trend is computed for the hydroxyl group dissociation at H2O or O covered surfaces. In contrast, the O–H bond breaking of water on oxygen covered Pd surfaces, H2Oad + Oad → 2OHad, is predicted to be likely with a barrier of ∼0.3 eV. The reverse reaction, 2OHad → H2Oad + Oad, is also found to be very feasible with a barrier of ∼0.1 eV. These results show that on oxygen-covered surfaces production of hydroxyl species is highly likely, supporting previous experimental findings.
Co-reporter:Zhe Li, Zhao-Xu Chen, Guo-Jun Kang, Xiang He
Catalysis Today (16 May 2011) Volume 165(Issue 1) pp:25-31
Publication Date(Web):16 May 2011
DOI:10.1016/j.cattod.2010.11.072
Density functional theory (DFT) calculations are performed to investigate the interactions of small Aun (n = 1–5) clusters with single-walled carbon nanotubes (CNTs) and H2 dissociation on the CNT supported clusters. Encapsulated Au clusters interact more strongly with the metallic CNTs than with the semi-conducting ones, where charge transfers from CNTs to the clusters play an important role. The clusters deposited outside the CNT are more stable than the ones encapsulated inside the tubes except for Au1 and Au3 on CNT(6, 6). Generally H2 dissociation becomes more favorable thermodynamically, especially on the encapsulated clusters. Except for Au monomer, H2 dissociation on the encapsulated clusters is kinetically more difficult than on the outside deposited clusters. Compared with the situation on bare clusters, H2 dissociation needs to overcome higher barriers on CNT supported clusters, apart from that on Au monomer on the outer surface of the CNTs and on the encapsulated Au dimer. These kinetic results demonstrate that confinement effect is not helpful for all reactions and CNT supported catalysts do not improve the activity for all reactions.
Co-reporter:Jiushuai Deng, Yanhua Lei, Shuming Wen, Zhaoxu Chen
International Journal of Mineral Processing (10 July 2015) Volume 140() pp:43-49
Publication Date(Web):10 July 2015
DOI:10.1016/j.minpro.2015.04.026
•Different valences and spin states significantly influence the interaction of copper and iron ions with ethyl xanthate.•High-spin Fe–EX complex with spin-V is the most stable.•Stronger interaction exists between copper (II) and xanthate ion compared with copper (I).The geometric and electronic structure properties of complexes of copper (Cu) and iron (Fe) ions with various valences and spin states with ethyl xanthate (EX) were calculated by the density functional theory (DFT/B3LYP). Analyses on the geometric parameters, interaction energies, natural bond orbital (NBO), and frontier molecular orbital reveal that there are two main structures in the metal–ethyl xanthate complexes, whose structure of metal atom with two sulfur atoms was more stable. Different spin status significantly influences the interaction of Fe ions with EX as well as the geometric structure of its complex. The most spin state for Fe2 + was spin-V, while it was spin-IV for Fe3 +. The binding energy of Fe3 + and EX is larger than that of Fe2 + and EX. Similarly, the binding energy of Cu2 + and EX is larger than that of Cu+ and EX. The covalent interaction between EX ion and Cu+ is relatively weak, whereas the ionic bond interaction is relatively strong. The solution effect hasn't been taken into account, but important information related to multi-spin states has been gained. The theoretical calculations in the present work may help explain the flotation mechanism and flotability difference of iron-bearing and copper-bearing minerals.
Co-reporter:Xiang He, Yucheng Huang and Zhao-Xu Chen
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 1) pp:NaN109-109
Publication Date(Web):2010/11/16
DOI:10.1039/C0CP01344G
Catalytic performances of alloy and surface alloy are sensitive to the surface structures and composition. In this paper we present an overall survey of the surface structure of Pd(111) covered with different amount of Zn using Monte Carlo simulations. We demonstrate that the composition of PdZn surface alloy is Zn coverage dependent: the surface concentration of Zn increases with the increase of the deposited Zn. At one or multi-layer of zinc deposited Pd(111), a multilayer 1:1 PdZn surface alloy will be formed. Surface alloy islands dominated by palladium are formed at submonolayer Zn coverage. At very low zinc coverage, small palladium ensembles of 3 to 5 Pd atoms exist preferentially on the Pd(111) surface. Our simulated results which are consistent with the pertinent experiments indicate that the unusual high-temperature desorption peak of formaldehyde observed experimentally has likely originated from the small surface ensembles induced by deposited Zn.
Co-reporter:Yunsheng Xue, Yuhui Wang, Zhongyan Cao, Jian Zhou and Zhao-Xu Chen
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 40) pp:NaN9597-9597
Publication Date(Web):2016/09/01
DOI:10.1039/C6OB01611A
Density functional theory (DFT) calculations were performed to elucidate the mechanism and the origin of the high enantioselectivity of the aza-Henry reaction of isatin-derived N-Boc ketimine catalyzed by a quinine-derived catalyst (QN). The C–C bond formation step is found to be both the rate-determining and the stereo-controlled step. The results revealed the important role of the phenolic OH group in pre-organizing the complex of nitromethane and QN and stabilizing the in situ-generated nitronate and protonated QN. Three possible activation modes for C–C bond formation involving different coordination patterns of catalyst and substrates were studied, and it was found that both the ion pair-hydrogen bonding mode and the Brønsted acid-hydrogen bonding mode are viable, with the latter slightly preferred for the real catalytic system. The calculated enantiomeric excess (ee) favouring the S enantiomer is in good agreement with the experimental result. The high reactivity and enantioselectivity can be ascribed to the cooperative role of the multiple non-covalent interactions, including classical and non-classical H bonding as well as anion⋯π interactions. These results also highlight the importance of the inclusion of dispersion correction for achieving a reasonable agreement between theory and experiment for the current reaction.
Co-reporter:Feng Cheng and Zhao-Xu Chen
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 5) pp:NaN3943-3943
Publication Date(Web):2015/12/22
DOI:10.1039/C5CP05020K
Pd/ZnO is a promising catalyst studied for methanol steam reforming (MSR) and the 1:1 PdZn alloy is demonstrated to be the active component. It is believed that MSR starts from methanol dehydrogenation to methoxy. Previous studies of methanol dehydrogenation on the ideal PdZn(111) surface show that methanol adsorbs weakly on the PdZn(111) surface and it is hard for methanol to transform into methoxy because of the high dehydrogenation barrier, indicating that the catalyst model is not appropriate for investigating the first step of MSR. Using the model derived from our recent kinetic Monte Carlo simulations, we examined the process CH3OH → CH3O → CH2O → CHO → CO. Compared with the ideal model, methanol adsorbs much more strongly and the barrier from CH3OH → CH3O is much lower on the kMC model. On the other hand, the C–H bond breaking of CH3O, CH2O and CHO becomes harder. We show that co-adsorbed water is important for refreshing the active sites. The present study shows that the first MSR step most likely takes place on three-fold hollow sites formed by Zn atoms, and the inhomogeneity of the PdZn alloy may exert significant influences on reactions.

![5,2,6-(Iminomethenimino)-1H-imidazo[4,5-b]pyrazine,octahydro-1,3,4,7,8,10-hexanitro-](http://img.cochemist.com/ccimg/135300/135285-90-4.png)
![5,2,6-(Iminomethenimino)-1H-imidazo[4,5-b]pyrazine,octahydro-1,3,4,7,8,10-hexanitro-](http://img.cochemist.com/ccimg/135300/135285-90-4_b.png)










![Pentacyclo[4.2.0.02,5.03,8.04,7]octane, 1,4-dinitro-](http://img.cochemist.com/ccimg/87900/87830-30-6.png)
![Pentacyclo[4.2.0.02,5.03,8.04,7]octane, 1,4-dinitro-](http://img.cochemist.com/ccimg/87900/87830-30-6_b.png)






















![6-Chlorobenzo[d]thiazole](http://img.cochemist.com/ccimg/3000/2942-10-1.png)
![6-Chlorobenzo[d]thiazole](http://img.cochemist.com/ccimg/3000/2942-10-1_b.png)