Co-reporter:Predrag Kukic, Yulia Pustovalova, Carlo Camilloni, Stefano Gianni, Dmitry M. Korzhnev, and Michele Vendruscolo
Journal of the American Chemical Society May 24, 2017 Volume 139(Issue 20) pp:6899-6899
Publication Date(Web):April 12, 2017
DOI:10.1021/jacs.7b01540
The nucleation–condensation mechanism represents a major paradigm to understand the folding process of many small globular proteins. Although substantial evidence has been acquired for this mechanism, it has remained very challenging to characterize the initial events leading to the formation of a folding nucleus. To achieve this goal, we used a combination of relaxation dispersion NMR spectroscopy and molecular dynamics simulations to determine ensembles of conformations corresponding to the denatured, transition, and native states in the folding of the activation domain of human procarboxypeptidase A2 (ADA2h). We found that the residues making up the folding nucleus tend to interact in the denatured state in a transient manner and not simultaneously, thereby forming incomplete and distorted versions of the folding nucleus. Only when all the contacts between these key residues are eventually formed can the protein reach the transition state and continue folding. Overall, our results elucidate the mechanism of formation of the folding nucleus of a protein and provide insights into how its folding rate can be modified during evolution by mutations that modulate the strength of the interactions between the residues forming the folding nucleus.
Co-reporter:Aditi N. Borkar;Pramodh Vallurupalli;Carlo Camilloni;Lewis E. Kay
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 4) pp:2797-2804
Publication Date(Web):2017/01/25
DOI:10.1039/C6CP08313G
RNA molecules in solution tend to undergo structural fluctuations of relatively large amplitude and to populate a range of different conformations some of which with low populations. It is still very challenging, however, to characterise the structures of these low populated states and to understand their functional roles. In the present study, we address this problem by using NMR residual dipolar couplings (RDCs) as structural restraints in replica-averaged metadynamics (RAM) simulations. By applying this approach to a 14-mer RNA hairpin containing the prototypical UUCG tetraloop motif, we show that it is possible to construct the free energy landscape of this RNA molecule. This free energy landscapes reveals the surprisingly rich dynamics of the UUCG tetraloop and identifies the multiple substates that exist in equilibrium owing to thermal fluctuations. The approach that we present is general and can be applied to the study of the free energy landscapes of other RNA or RNA-protein systems.
Co-reporter:Gabriella T. Heller, Francesco A. Aprile, Massimiliano Bonomi, Carlo Camilloni, ... Michele Vendruscolo
Journal of Molecular Biology 2017 Volume 429, Issue 18(Volume 429, Issue 18) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.jmb.2017.07.016
•We show that a small molecule binds a disordered peptide in a diffuse, entropic manner.•We observe sequence specificity in the diffuse binding, suggesting that specificity in this type of binding may be achievable.•We demonstrate that this type of binding can alter the biophysical properties of a disordered peptide.Approximately one-third of the human proteome is made up of proteins that are entirely disordered or that contain extended disordered regions. Although these disordered proteins are closely linked with many major diseases, their binding mechanisms with small molecules remain poorly understood, and a major concern is whether their specificity can be sufficient for drug development. Here, by studying the interaction of a small molecule and a disordered peptide from the oncogene protein c-Myc, we describe a “specific-diffuse” binding mechanism that exhibits sequence specificity despite being of entropic nature. By combining NMR spectroscopy, biophysical measurements, statistical inference, and molecular simulations, we provide a quantitative measure of such sequence specificity and compare it to the case of the interaction of urea, which is diffuse but not specific. To investigate whether this type of binding can generally modify intermolecular interactions, we show that it leads to an inhibition of the aggregation of the peptide. These results suggest that the binding mechanism that we report may create novel opportunities to discover drugs that target disordered proteins in their monomeric states in a specific manner.Download high-res image (279KB)Download full-size image
Co-reporter:Sean Chia;Patrick Flagmeier;Johnny Habchi;Veronica Lattanzi;Sara Linse;Christopher M. Dobson;Tuomas P. J. Knowles
PNAS 2017 Volume 114 (Issue 30 ) pp:8005-8010
Publication Date(Web):2017-07-25
DOI:10.1073/pnas.1700239114
The coaggregation of the amyloid-β peptide (Aβ) and α-synuclein is commonly observed in a range of neurodegenerative disorders,
including Alzheimer’s and Parkinson’s diseases. The complex interplay between Aβ and α-synuclein has led to seemingly contradictory
results on whether α-synuclein promotes or inhibits Aβ aggregation. Here, we show how these conflicts can be rationalized
and resolved by demonstrating that different structural forms of α-synuclein exert different effects on Aβ aggregation. Our
results demonstrate that whereas monomeric α-synuclein blocks the autocatalytic proliferation of Aβ42 (the 42-residue form
of Aβ) fibrils, fibrillar α-synuclein catalyses the heterogeneous nucleation of Aβ42 aggregates. It is thus the specific balance
between the concentrations of monomeric and fibrillar α-synuclein that determines the outcome of the Aβ42 aggregation reaction.
Co-reporter:Massimiliano Bonomi, Gabriella T. Heller, Carlo Camilloni, Michele Vendruscolo
Current Opinion in Structural Biology 2017 Volume 42(Volume 42) pp:
Publication Date(Web):1 February 2017
DOI:10.1016/j.sbi.2016.12.004
•The principles of protein structural ensemble determination are described.•The use of experimental data averaged over multiple states is analysed.•Approaches for accounting for errors in the data and models are presented.•Major questions about protein structural ensemble determination are summarised.•Community goals for the study of disordered proteins are discussed.The biological functions of protein molecules are intimately dependent on their conformational dynamics. This aspect is particularly evident for disordered proteins, which constitute perhaps one-third of the human proteome. Therefore, structural ensembles often offer more useful representations of proteins than individual conformations. Here, we describe how the well-established principles of protein structure determination should be extended to the case of protein structural ensembles determination. These principles concern primarily how to deal with conformationally heterogeneous states, and with experimental measurements that are averaged over such states and affected by a variety of errors. We first review the growing literature of recent methods that combine experimental and computational information to model structural ensembles, highlighting their similarities and differences. We then address some conceptual problems in the determination of structural ensembles and define future goals towards the establishment of objective criteria for the comparison, validation, visualization and dissemination of such ensembles.
Co-reporter:Gabriella T. Heller;Francesco A. Aprile
Cellular and Molecular Life Sciences 2017 Volume 74( Issue 17) pp:3225-3243
Publication Date(Web):19 June 2017
DOI:10.1007/s00018-017-2563-4
It is generally recognized that a large fraction of the human proteome is made up of proteins that remain disordered in their native states. Despite the fact that such proteins play key biological roles and are involved in many major human diseases, they still represent challenging targets for drug discovery. A major bottleneck for the identification of compounds capable of interacting with these proteins and modulating their disease-promoting behaviour is the development of effective techniques to probe such interactions. The difficulties in carrying out binding measurements have resulted in a poor understanding of the mechanisms underlying these interactions. In order to facilitate further methodological advances, here we review the most commonly used techniques to probe three types of interactions involving small molecules: (1) those that disrupt functional interactions between disordered proteins; (2) those that inhibit the aberrant aggregation of disordered proteins, and (3) those that lead to binding disordered proteins in their monomeric states. In discussing these techniques, we also point out directions for future developments.
Co-reporter:Predrag Kukic, Patrik Lundström, Carlo Camilloni, Johan Evenäs, Mikael Akke, and Michele Vendruscolo
Biochemistry 2016 Volume 55(Issue 1) pp:19-28
Publication Date(Web):November 30, 2015
DOI:10.1021/acs.biochem.5b00961
Calmodulin is a two-domain signaling protein that becomes activated upon binding cooperatively two pairs of calcium ions, leading to large-scale conformational changes that expose its binding site. Despite significant advances in understanding the structural biology of calmodulin functions, the mechanistic details of the conformational transition between closed and open states have remained unclear. To investigate this transition, we used a combination of molecular dynamics simulations and nuclear magnetic resonance (NMR) experiments on the Ca2+-saturated E140Q C-terminal domain variant. Using chemical shift restraints in replica-averaged metadynamics simulations, we obtained a high-resolution structural ensemble consisting of two conformational states and validated such an ensemble against three independent experimental data sets, namely, interproton nuclear Overhauser enhancements, 15N order parameters, and chemical shift differences between the exchanging states. Through a detailed analysis of this structural ensemble and of the corresponding statistical weights, we characterized a calcium-mediated conformational transition whereby the coordination of Ca2+ by just one oxygen of the bidentate ligand E140 triggers a concerted movement of the two EF-hands that exposes the target binding site. This analysis provides atomistic insights into a possible Ca2+-mediated activation mechanism of calmodulin that cannot be achieved from static structures alone or from ensemble NMR measurements of the transition between conformations.
Co-reporter:Priyanka Joshi, Sean Chia, Johnny Habchi, Tuomas P. J. Knowles, Christopher M. Dobson, and Michele Vendruscolo
ACS Combinatorial Science 2016 Volume 18(Issue 3) pp:144
Publication Date(Web):February 29, 2016
DOI:10.1021/acscombsci.5b00129
The aggregation process of intrinsically disordered proteins (IDPs) has been associated with a wide range of neurodegenerative disorders, including Alzheimer’s and Parkinson’s diseases. Currently, however, no drug in clinical use targets IDP aggregation. To facilitate drug discovery programs in this important and challenging area, we describe a fragment-based approach of generating small-molecule libraries that target specific IDPs. The method is based on the use of molecular fragments extracted from compounds reported in the literature to inhibit of the aggregation of IDPs. These fragments are used to screen existing large generic libraries of small molecules to form smaller libraries specific for given IDPs. We illustrate this approach by describing three distinct small-molecule libraries to target, Aβ, tau, and α-synuclein, which are three IDPs implicated in Alzheimer’s and Parkinson’s diseases. The strategy described here offers novel opportunities for the identification of effective molecular scaffolds for drug discovery for neurodegenerative disorders and to provide insights into the mechanism of small-molecule binding to IDPs.Keywords: Alzheimer’s disease; drug discovery; Parkinson’s disease; protein aggregation
Co-reporter:Farah El-Turk, Francisco N. Newby, Erwin De Genst, Tim Guilliams, Tara Sprules, Anthony Mittermaier, Christopher M. Dobson, and Michele Vendruscolo
Biochemistry 2016 Volume 55(Issue 22) pp:3116-3122
Publication Date(Web):April 20, 2016
DOI:10.1021/acs.biochem.6b00149
α-Synuclein is an intrinsically disordered protein whose aggregation is associated with Parkinson’s disease and other related neurodegenerative disorders. Recently, two single-domain camelid antibodies (nanobodies) were shown to bind α-synuclein with high affinity. Herein, we investigated how these two nanobodies (NbSyn2 and NbSyn87), which are directed to two distinct epitopes within the C-terminal domain of α-synuclein, affect the conformational properties of this protein. Our results suggest that nanobody NbSyn2, which binds to the five C-terminal residues of α-synuclein (residues 136–140), does not disrupt the transient long-range interactions that generate a degree of compaction within the native structural ensemble of α-synuclein. In contrast, the data that we report indicate that NbSyn87, which targets a central region within the C-terminal domain (residues 118–128), has more substantial effects on the fluctuating secondary and tertiary structure of the protein. These results are consistent with the different effects that the two nanobodies have on the aggregation behavior of α-synuclein in vitro. Our findings thus provide new insights into the type of effects that nanobodies can have on the conformational ensemble of α-synuclein.
Co-reporter:Rosie Freer;Pietro Sormanni;Giulia Vecchi;Prajwal Ciryam;Christopher M. Dobson
Science Advances 2016 Vol 2(8) pp:e1600947
Publication Date(Web):10 Aug 2016
DOI:10.1126/sciadv.1600947
A tissue vulnerability map based on Aβ and tau homeostasis in healthy brains predicts the staging of Alzheimer’s disease.
Co-reporter:Massimiliano Bonomi;Carlo Camilloni;Andrea Cavalli
Science Advances 2016 Volume 2(Issue 1) pp:e1501177
Publication Date(Web):22 Jan 2016
DOI:10.1126/sciadv.1501177
Researchers present a Bayesian inference method for heterogeneous systems that integrates prior information with noisy experimental data.
Co-reporter:Prajwal Ciryam;Rishika Kundra;Rosie Freer;Richard I. Morimoto;Christopher M. Dobson
PNAS 2016 Volume 113 (Issue 17 ) pp:4753-4758
Publication Date(Web):2016-04-26
DOI:10.1073/pnas.1516604113
It is well-established that widespread transcriptional changes accompany the onset and progression of Alzheimer’s disease.
Because of the multifactorial nature of this neurodegenerative disorder and its complex relationship with aging, however,
it remains unclear whether such changes are the result of nonspecific dysregulation and multisystem failure or instead are
part of a coordinated response to cellular dysfunction. To address this problem in a systematic manner, we performed a meta-analysis
of about 1,600 microarrays from human central nervous system tissues to identify transcriptional changes upon aging and as
a result of Alzheimer’s disease. Our strategy to discover a transcriptional signature of Alzheimer’s disease revealed a set
of down-regulated genes that encode proteins metastable to aggregation. Using this approach, we identified a small number
of biochemical pathways, notably oxidative phosphorylation, enriched in proteins vulnerable to aggregation in control brains
and encoded by genes down-regulated in Alzheimer’s disease. These results suggest that the down-regulation of a metastable
subproteome may help mitigate aberrant protein aggregation when protein homeostasis becomes compromised in Alzheimer’s disease.
Co-reporter:Aditi N. Borkar;Carlo Camilloni;Michael F. Bardaro, Jr.;Francesco A. Aprile;Gabriele Varani
PNAS 2016 Volume 113 (Issue 26 ) pp:7171-7176
Publication Date(Web):2016-06-28
DOI:10.1073/pnas.1521349113
The interaction of the HIV-1 protein transactivator of transcription (Tat) and its cognate transactivation response element
(TAR) RNA transactivates viral transcription and represents a paradigm for the widespread occurrence of conformational rearrangements
in protein-RNA recognition. Although the structures of free and bound forms of TAR are well characterized, the conformations
of the intermediates in the binding process are still unknown. By determining the free energy landscape of the complex using
NMR residual dipolar couplings in replica-averaged metadynamics simulations, we observe two low-population intermediates.
We then rationally design two mutants, one in the protein and another in the RNA, that weaken specific nonnative interactions
that stabilize one of the intermediates. By using surface plasmon resonance, we show that these mutations lower the release
rate of Tat, as predicted. These results identify the structure of an intermediate for RNA-protein binding and illustrate
a general strategy to achieve this goal with high resolution.
Co-reporter:Francisco N. Newby, Alfonso De Simone, Maho Yagi-Utsumi, Xavier Salvatella, Christopher M. Dobson, and Michele Vendruscolo
Biochemistry 2015 Volume 54(Issue 46) pp:6876-6886
Publication Date(Web):October 19, 2015
DOI:10.1021/acs.biochem.5b00670
Residual dipolar couplings (RDCs) and paramagnetic relaxation enhancements (PREs) have emerged as valuable parameters for defining the structures and dynamics of disordered proteins by nuclear magnetic resonance (NMR) spectroscopy. Procedures for their measurement, however, may lead to conformational perturbations because of the presence of the alignment media necessary for recording RDCs, or of the paramagnetic groups that must be introduced for measuring PREs. We discuss here experimental methods for quantifying these effects by considering the case of the 40-residue isoform of the amyloid β peptide (Aβ40), which is associated with Alzheimer’s disease. By conducting RDC measurements over a range of concentrations of certain alignment media, we show that perturbations arising from transient binding of Aβ40 can be characterized, allowing appropriate corrections to be made. In addition, by using NMR experiments sensitive to long-range interactions, we show that it is possible to identify relatively nonperturbing sites for attaching nitroxide radicals for PRE measurements. Thus, minimizing the conformational perturbations introduced by RDC and PRE measurements should facilitate their use for the rigorous determination of the conformational properties of disordered proteins.
Co-reporter:Francesco A. Aprile, Pietro Sormanni, and Michele Vendruscolo
Biochemistry 2015 Volume 54(Issue 32) pp:5103-5112
Publication Date(Web):July 20, 2015
DOI:10.1021/acs.biochem.5b00459
Molecular chaperones facilitate the folding and assembly of proteins and inhibit their aberrant aggregation. They thus offer several opportunities for biomedical and biotechnological applications, as for example they can often prevent protein aggregation more effectively than other therapeutic molecules, including small molecules and antibodies. Here we present a method of designing molecular chaperones with enhanced activity against specific amyloidogenic substrates while leaving unaltered their functions toward other substrates. The method consists of grafting onto a molecular chaperone a peptide designed to bind specifically an epitope in the target substrate. We illustrate this strategy by describing Hsp70 variants with increased affinities for α-synuclein and Aβ42 but otherwise unaltered affinities for other substrates. These designed variants inhibit protein aggregation and disaggregate preformed fibrils significantly more effectively than wild-type Hsp70 indicating that the strategy presented here provides a possible route for tailoring rationally molecular chaperones for specific purposes.
Co-reporter:Carlo Camilloni and Michele Vendruscolo
Biochemistry 2015 Volume 54(Issue 51) pp:7470-7476
Publication Date(Web):December 1, 2015
DOI:10.1021/acs.biochem.5b01138
Nuclear magnetic resonance (NMR) spectroscopy provides detailed information about the structure and dynamics of proteins by exploiting the conformational dependence of the magnetic properties of certain atomic nuclei. The mapping between NMR measurements and molecular structures, however, often requires approximated descriptions based on the fitting of a number of parameters, thus reducing the quality of the information available from the experiments. To improve on this limitation, we show here that it is possible to use pseudocontact shifts and residual dipolar couplings as “exact” NMR restraints. We implement this strategy by using a replica-averaging method and illustrate its application by calculating an ensemble of structures representing the dynamics of the two-domain protein calmodulin.
Co-reporter:Pietro Sormanni;Francesco A. Aprile
PNAS 2015 Volume 112 (Issue 32 ) pp:9902-9907
Publication Date(Web):2015-08-11
DOI:10.1073/pnas.1422401112
Antibodies are powerful tools in life sciences research, as well as in diagnostic and therapeutic applications, because of
their ability to bind given molecules with high affinity and specificity. Using current methods, however, it is laborious
and sometimes difficult to generate antibodies to target specific epitopes within a protein, in particular if these epitopes
are not effective antigens. Here we present a method to rationally design antibodies to enable them to bind virtually any
chosen disordered epitope in a protein. The procedure consists in the sequence-based design of one or more complementary peptides
targeting a selected disordered epitope and the subsequent grafting of such peptides on an antibody scaffold. We illustrate
the method by designing six single-domain antibodies to bind different epitopes within three disease-related intrinsically
disordered proteins and peptides (α-synuclein, Aβ42, and IAPP). Our results show that all these designed antibodies bind their
targets with good affinity and specificity. As an example of an application, we show that one of these antibodies inhibits
the aggregation of α-synuclein at substoichiometric concentrations and that binding occurs at the selected epitope. Taken
together, these results indicate that the design strategy that we propose makes it possible to obtain antibodies targeting
given epitopes in disordered proteins or protein regions.
Co-reporter:Carlo Camilloni and Michele Vendruscolo
The Journal of Physical Chemistry B 2015 Volume 119(Issue 3) pp:653-661
Publication Date(Web):May 14, 2014
DOI:10.1021/jp5021824
Residual dipolar couplings (RDCs) are parameters measured in nuclear magnetic resonance spectroscopy that can provide exquisitely detailed information about the structure and dynamics of biological macromolecules. We describe here a method of using RDCs for the structural and dynamical refinement of proteins that is based on the observation that the RDC between two atomic nuclei depends directly on the angle ϑ between the internuclear vector and the external magnetic field. For every pair of nuclei for which an RDC is available experimentally, we introduce a structural restraint to minimize the deviation from the value of the angle ϑ derived from the measured RDC and that calculated in the refinement protocol. As each restraint involves only the calculation of the angle ϑ of the corresponding internuclear vector, the method does not require the definition of an overall alignment tensor to describe the preferred orientation of the protein with respect to the alignment medium. Application to the case of ubiquitin demonstrates that this method enables an accurate refinement of the structure and dynamics of this protein to be obtained.
Co-reporter:Carlo Camilloni
Journal of the American Chemical Society 2014 Volume 136(Issue 25) pp:8982-8991
Publication Date(Web):June 2, 2014
DOI:10.1021/ja5027584
The characterization of denatured states of proteins is challenging because the lack of permanent structure in these states makes it difficult to apply to them standard methods of structural biology. In this work we use all-atom replica-averaged metadynamics (RAM) simulations with NMR chemical shift restraints to determine an ensemble of structures representing an acid-denatured state of the 86-residue protein ACBP. This approach has enabled us to reach convergence in the free energy landscape calculations, obtaining an ensemble of structures in relatively accurate agreement with independent experimental data used for validation. By observing at atomistic resolution the transient formation of native and non-native structures in this acid-denatured state of ACBP, we rationalize the effects of single-point mutations on the folding rate, stability, and transition-state structures of this protein, thus characterizing the role of the unfolded state in determining the folding process.
Co-reporter:Jane R. Allison, Robert C. Rivers, John C. Christodoulou, Michele Vendruscolo, and Christopher M. Dobson
Biochemistry 2014 Volume 53(Issue 46) pp:
Publication Date(Web):October 28, 2014
DOI:10.1021/bi5009326
α-Synuclein is an intrinsically disordered protein whose aggregation is implicated in Parkinson’s disease. A second member of the synuclein family, β-synuclein, shares significant sequence similarity with α-synuclein but is much more resistant to aggregation. β-Synuclein is missing an 11-residue stretch in the central non-β-amyloid component region that forms the core of α-synuclein amyloid fibrils, yet insertion of these residues into β-synuclein to produce the βSHC construct does not markedly increase the aggregation propensity. To investigate the structural basis of these different behaviors, quantitative nuclear magnetic resonance data, in the form of paramagnetic relaxation enhancement-derived interatomic distances, are combined with molecular dynamics simulations to generate ensembles of structures representative of the solution states of α-synuclein, β-synuclein, and βSHC. Comparison of these ensembles reveals that the differing aggregation propensities of α-synuclein and β-synuclein are associated with differences in the degree of residual structure in the C-terminus coupled to the shorter separation between the N- and C-termini in β-synuclein and βSHC, making protective intramolecular contacts more likely.
Co-reporter:Carlo Camilloni;Aleksandr B. Sahakyan;Michael J. Holliday;Nancy G. Isern;Fengli Zhang;Elan Z. Eisenmesser
PNAS 2014 Volume 111 (Issue 28 ) pp:10203-10208
Publication Date(Web):2014-07-15
DOI:10.1073/pnas.1404220111
Proline isomerization is a ubiquitous process that plays a key role in the folding of proteins and in the regulation of their
functions. Different families of enzymes, known as “peptidyl-prolyl isomerases” (PPIases), catalyze this reaction, which involves
the interconversion between the cis and trans isomers of the N-terminal amide bond of the amino acid proline. However, complete descriptions of the mechanisms by which
these enzymes function have remained elusive. We show here that cyclophilin A, one of the most common PPIases, provides a
catalytic environment that acts on the substrate through an electrostatic handle mechanism. In this mechanism, the electrostatic
field in the catalytic site turns the electric dipole associated with the carbonyl group of the amino acid preceding the proline
in the substrate, thus causing the rotation of the peptide bond between the two residues. We identified this mechanism using
a combination of NMR measurements, molecular dynamics simulations, and density functional theory calculations to simultaneously
determine the cis-bound and trans-bound conformations of cyclophilin A and its substrate as the enzymatic reaction takes place. We anticipate that this approach
will be helpful in elucidating whether the electrostatic handle mechanism that we describe here is common to other PPIases
and, more generally, in characterizing other enzymatic processes.
Co-reporter:Pengfei Tian;Jesper Ferkinghoff-Borg;Wouter Boomsma;Jes Frellsen;Kresten Lindorff-Larsen;Thomas Hamelryck
PNAS 2014 Volume 111 (Issue 38 ) pp:13852-13857
Publication Date(Web):2014-09-23
DOI:10.1073/pnas.1404948111
Methods of protein structure determination based on NMR chemical shifts are becoming increasingly common. The most widely
used approaches adopt the molecular fragment replacement strategy, in which structural fragments are repeatedly reassembled
into different complete conformations in molecular simulations. Although these approaches are effective in generating individual
structures consistent with the chemical shift data, they do not enable the sampling of the conformational space of proteins
with correct statistical weights. Here, we present a method of molecular fragment replacement that makes it possible to perform
equilibrium simulations of proteins, and hence to determine their free energy landscapes. This strategy is based on the encoding
of the chemical shift information in a probabilistic model in Markov chain Monte Carlo simulations. First, we demonstrate
that with this approach it is possible to fold proteins to their native states starting from extended structures. Second,
we show that the method satisfies the detailed balance condition and hence it can be used to carry out an equilibrium sampling
from the Boltzmann distribution corresponding to the force field used in the simulations. Third, by comparing the results
of simulations carried out with and without chemical shift restraints we describe quantitatively the effects that these restraints
have on the free energy landscapes of proteins. Taken together, these results demonstrate that the molecular fragment replacement
strategy can be used in combination with chemical shift information to characterize not only the native structures of proteins
but also their conformational fluctuations.
Co-reporter:Arvind Kannan ; Carlo Camilloni ; Aleksandr B. Sahakyan ; Andrea Cavalli
Journal of the American Chemical Society 2013 Volume 136(Issue 6) pp:2204-2207
Publication Date(Web):December 31, 2013
DOI:10.1021/ja4105396
Recent improvements in the accuracy of structure-based methods for the prediction of nuclear magnetic resonance chemical shifts have inspired numerous approaches for determining the secondary and tertiary structures of proteins. Such advances also suggest the possibility of using chemical shifts to characterize the conformational fluctuations of these molecules. Here we describe a method of using methyl chemical shifts as restraints in replica-averaged molecular dynamics (MD) simulations, which enables us to determine the conformational ensemble of the HU dimer and characterize the range of motions accessible to its flexible β-arms. Our analysis suggests that the bending action of HU on DNA is mediated by a mechanical clamping mechanism, in which metastable structural intermediates sampled during the hinge motions of the β-arms in the free state are presculpted to bind DNA. These results illustrate that using side-chain chemical shift data in conjunction with MD simulations can provide quantitative information about the free energy landscapes of proteins and yield detailed insights into their functional mechanisms.
Co-reporter:Carlo Camilloni, Andrea Cavalli, and Michele Vendruscolo
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 12) pp:5610-5617
Publication Date(Web):November 5, 2013
DOI:10.1021/ct4006272
A statistical mechanics description of complex molecular systems involves the determination of ensembles of conformations that represent their Boltzmann distributions. The observable properties of these systems can be then predicted by calculating averages over such ensembles. In principle, given accurate energy functions and efficient sampling methods, these ensembles can be generated by molecular dynamics simulations. In practice, however, often the energy functions are known only approximately and the sampling can be carried out only in a limited manner. We describe here a method that enables to increase simultaneously both the quality of the energy functions and of the extent of the sampling in a system-dependent manner. The method is based on the incorporation of experimental data as replica-averaged structural restraints in molecular dynamics simulations and exploits the metadynamics framework to enhance the sampling. The application to the case of α-conotoxin SI, a 13-residue peptide that has been characterized extensively by experimental measurements, shows that the approach that we describe enables accurate free energy landscapes to be generated. The analysis of these landscapes indicates the presence of a low population state in equilibrium with the native state in which the only aromatic residue of α-conotoxin SI is exposed to the solvent, which is a feature that may predispose the peptide to interact with its partners.
Co-reporter:Alfonso De Simone, Rinaldo W. Montalvao, Christopher M. Dobson, and Michele Vendruscolo
Biochemistry 2013 Volume 52(Issue 37) pp:
Publication Date(Web):August 14, 2013
DOI:10.1021/bi4007513
Hen lysozyme is an enzyme characterized by the presence of two domains whose relative motions are involved in the mechanism of binding and release of the substrates. By using residual dipolar couplings as replica-averaged structural restraints in molecular dynamics simulations, we characterize the breathing motions describing the interdomain fluctuations of this protein. We found that the ensemble of conformations that we determined spans the entire range of structures of hen lysozyme deposited in the Protein Data Bank, including both the free and bound states, suggesting that the thermal motions in the free state provide access to the structures populated upon binding. The approach that we present illustrates how the use of residual dipolar couplings as replica-averaged structural restraints in molecular dynamics simulations makes it possible to explore conformational fluctuations of a relatively large amplitude in proteins.
Co-reporter:Alfonso De Simone, Martin Gustavsson, Rinaldo W. Montalvao, Lei Shi, Gianluigi Veglia, and Michele Vendruscolo
Biochemistry 2013 Volume 52(Issue 38) pp:
Publication Date(Web):August 22, 2013
DOI:10.1021/bi400517b
Phospholamban is an integral membrane protein that controls the calcium balance in cardiac muscle cells. As the function and regulation of this protein require the active involvement of low populated states in equilibrium with the native state, it is of great interest to acquire structural information about them. In this work, we calculate the conformations and populations of the ground state and the three main excited states of phospholamban by incorporating nuclear magnetic resonance residual dipolar couplings as replica-averaged structural restraints in molecular dynamics simulations. We then provide a description of the manner in which phosphorylation at Ser16 modulates the activity of the protein by increasing the sizes of the populations of its excited states. These results demonstrate that the approach that we describe provides a detailed characterization of the different states of phospholamban that determine the function and regulation of this membrane protein. We anticipate that the knowledge of conformational ensembles enable the design of new dominant negative mutants of phospholamban by modulating the relative populations of its conformational substates.
Co-reporter:Carlo Camilloni and Michele Vendruscolo
The Journal of Physical Chemistry B 2013 Volume 117(Issue 37) pp:10737-10741
Publication Date(Web):August 13, 2013
DOI:10.1021/jp405614j
Intrinsically disordered proteins constitute a significant part of the human proteome and carry out a wide range of different functions, including in particular signaling and regulation. Several of these proteins are vulnerable to aggregation, and their aberrant assemblies have been associated with a variety of neurodegenerative and systemic diseases. It remains unclear, however, the extent to which the conformational properties of intrinsically disordered proteins in their monomeric states influence the aggregation behavior of these molecules. Here we report a relationship between aggregation rates and secondary structure populations in the soluble monomeric states of a series of mutational variants of α-synuclein. Overall, we found a correlation of over 90% between the changes in β-sheet populations calculated from NMR chemical shift data and the changes in aggregation rates for eight human-to-mouse chimeric mutants. These results provide support to the idea of investigating therapeutic strategies based on the stabilization of the monomeric form of intrinsically disordered proteins through the alteration of their conformational properties.
Co-reporter:Aleksandr B. Sahakyan and Michele Vendruscolo
The Journal of Physical Chemistry B 2013 Volume 117(Issue 7) pp:1989-1998
Publication Date(Web):February 11, 2013
DOI:10.1021/jp3057306
Ring current and electric field effects can considerably influence NMR chemical shifts in biomolecules. Understanding such effects is particularly important for the development of accurate mappings between chemical shifts and the structures of nucleic acids. In this work, we first analyzed the Pople and the Haigh–Mallion models in terms of their ability to describe nitrogen base conjugated ring effects. We then created a database (DiBaseRNA) of three-dimensional arrangements of RNA base pairs from X-ray structures, calculated the corresponding chemical shifts via a hybrid density functional theory approach and used the results to parametrize the ring current and electric field effects in RNA bases. Next, we studied the coupling of the electric field and ring current effects for different inter-ring arrangements found in RNA bases using linear model fitting, with joint electric field and ring current, as well as only electric field and only ring current approximations. Taken together, our results provide a characterization of the interdependence of ring current and electric field geometric factors, which is shown to be especially important for the chemical shifts of non-hydrogen atoms in RNA bases.
Co-reporter:Carlo Camilloni ; Paul Robustelli ; Alfonso De Simone ; Andrea Cavalli
Journal of the American Chemical Society 2012 Volume 134(Issue 9) pp:3968-3971
Publication Date(Web):February 9, 2012
DOI:10.1021/ja210951z
Following the recognition that NMR chemical shifts can be used for protein structure determination, rapid advances have recently been made in methods for extending this strategy for proteins and protein complexes of increasing size and complexity. A remaining major challenge is to develop approaches to exploit the information contained in the chemical shifts about conformational fluctuations in native states of proteins. In this work we show that it is possible to determine an ensemble of conformations representing the free energy surface of RNase A using chemical shifts as replica-averaged restraints in molecular dynamics simulations. Analysis of this surface indicates that chemical shifts can be used to characterize the conformational equilibrium between the two major substates of this protein.
Co-reporter:Carlo Camilloni, Alfonso De Simone, Wim F. Vranken, and Michele Vendruscolo
Biochemistry 2012 Volume 51(Issue 11) pp:
Publication Date(Web):February 23, 2012
DOI:10.1021/bi3001825
One of the major open challenges in structural biology is to achieve effective descriptions of disordered states of proteins. This problem is difficult because these states are conformationally highly heterogeneous and cannot be represented as single structures, and therefore it is necessary to characterize their conformational properties in terms of probability distributions. Here we show that it is possible to obtain highly quantitative information about particularly important types of probability distributions, the populations of secondary structure elements (α-helix, β-strand, random coil, and polyproline II), by using the information provided by backbone chemical shifts. The application of this approach to mammalian prions indicates that for these proteins a key role in molecular recognition is played by disordered regions characterized by highly conserved polyproline II populations. We also determine the secondary structure populations of a range of other disordered proteins that are medically relevant, including p53, α-synuclein, and the Aβ peptide, as well as an oligomeric form of αB-crystallin. Because chemical shifts are the nuclear magnetic resonance parameters that can be measured under the widest variety of conditions, our approach can be used to obtain detailed information about secondary structure populations for a vast range of different protein states.
Co-reporter:Cintia Roodveldt, August Andersson, Erwin J. De Genst, Adahir Labrador-Garrido, Alexander K. Buell, Christopher M. Dobson, Gian Gaetano Tartaglia, and Michele Vendruscolo
Biochemistry 2012 Volume 51(Issue 44) pp:
Publication Date(Web):September 24, 2012
DOI:10.1021/bi300558q
The aggregation process of α-synuclein, a protein closely associated with Parkinson’s disease, is highly sensitive to sequence variations. It is therefore of great importance to understand the factors that define the aggregation propensity of specific mutational variants as well as their toxic behavior in the cellular environment. In this context, we investigated the extent to which the aggregation behavior of α-synuclein can be altered to resemble that of β-synuclein, an aggregation-resistant homologue of α-synuclein not associated with disease, by swapping residues between the two proteins. Because of the vast number of possible swaps, we have applied a rational design procedure to single out a mutational variant, called α2β, in which two short stretches of the sequence in the NAC region have been replaced in α-synuclein from β-synuclein. We find not only that the aggregation rate of α2β is close to that of β-synuclein, being much lower than that of α-synuclein, but also that α2β effectively changes the cellular toxicity of α-synuclein to a value similar to that of β-synuclein upon exposure of SH-SY5Y cells to preformed oligomers. Remarkably, control experiments on the corresponding mutational variant of β-synuclein, called β2α, confirmed that the mutations that we have identified also shift the aggregation behavior of this protein toward that of α-synuclein. These results demonstrate that it is becoming possible to control in quantitative detail the sequence code that defines the aggregation behavior and toxicity of α-synuclein.
Co-reporter:Aleksandr B. Sahakyan, Andrea Cavalli, Wim F. Vranken, and Michele Vendruscolo
The Journal of Physical Chemistry B 2012 Volume 116(Issue 16) pp:4754-4759
Publication Date(Web):March 28, 2012
DOI:10.1021/jp2122054
We present a method of assessing the quality of protein structures based on the use of side-chain NMR chemical shifts. Because these parameters are very accurate reporters of side-chain positions and are highly sensitive to tertiary structure and packing, they are particularly useful for structure validation. To analyze a given structure, we define a quality score, QCS, that compares the chemical shifts calculated from such a structure with the corresponding experimental values in a way that takes account of the errors in the calculations. The results that we report illustrate the advantages in the examination of the quality of protein structures from the perspective of side-chains.
Co-reporter:Alfonso De Simone, Rinaldo W. Montalvao, and Michele Vendruscolo
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 12) pp:4189-4195
Publication Date(Web):October 10, 2011
DOI:10.1021/ct200361b
In order to carry out their functions, proteins often undergo significant conformational fluctuations that enable them to interact with their partners. The accurate characterization of these motions is key in order to understand the mechanisms by which macromolecular recognition events take place. Nuclear magnetic resonance spectroscopy offers a variety of powerful methods to achieve this result. We discuss a method of using residual dipolar couplings as replica-averaged restraints in molecular dynamics simulations to determine large amplitude motions of proteins, including those involved in the conformational equilibria that are established through interconversions between different states. By applying this method to ribonuclease A, we show that it enables one to characterize the ample fluctuations in interdomain orientations expected to play an important functional role.
Co-reporter:Kai J. Kohlhoff, Thomas R. Jahn, David A. Lomas, Christopher M. Dobson, Damian C. Crowther and Michele Vendruscolo
Integrative Biology 2011 vol. 3(Issue 7) pp:755-760
Publication Date(Web):23 Jun 2011
DOI:10.1039/C0IB00149J
The use of animal models in medical research provides insights into molecular and cellular mechanisms of human disease, and helps identify and test novel therapeutic strategies. Drosophila melanogaster—the common fruit fly—is one of the most well-established model organisms, as its study can be performed more readily and with far less expense than for other model animal systems, such as mice, fish, or primates. In the case of fruit flies, standard assays are based on the analysis of longevity and basic locomotor functions. Here we present the iFly tracking system, which enables to increase the amount of quantitative information that can be extracted from these studies, and to reduce significantly the duration and costs associated with them. The iFly system uses a single camera to simultaneously track the trajectories of up to 20 individual flies with about 100 μm spatial and 33 ms temporal resolution. The statistical analysis of fly movements recorded with such accuracy makes it possible to perform a rapid and fully automated quantitative analysis of locomotor changes in response to a range of different stimuli. We anticipate that the iFly method will reduce very considerably the costs and the duration of the testing of genetic and pharmacological interventions in Drosophila models, including an earlier detection of behavioural changes and a large increase in throughput compared to current longevity and locomotor assays.
Co-reporter:Andrea Cavalli, Rinaldo W. Montalvao, and Michele Vendruscolo
The Journal of Physical Chemistry B 2011 Volume 115(Issue 30) pp:9491-9494
Publication Date(Web):June 3, 2011
DOI:10.1021/jp202647q
Methods for determining protein structures using only chemical shift information are progressively becoming more accurate and reliable. A major problem, however, in the use of chemical shifts for the determination of the structures of protein complexes is that the changes in the chemical shifts upon binding tend to be rather limited and indeed often smaller than the standard errors made in the predictions of chemical shifts corresponding to given structures. We present a procedure that, despite this problem, enables one to use of chemical shifts to determine accurately the conformational changes that take place upon complex formation.
Co-reporter:Aleksr B. Sahakyan;Dr. Wim F. Vranken;Dr. Andrea Cavalli; Michele Vendruscolo
Angewandte Chemie International Edition 2011 Volume 50( Issue 41) pp:9620-9623
Publication Date(Web):
DOI:10.1002/anie.201101641
Co-reporter:Aleksr B. Sahakyan;Dr. Wim F. Vranken;Dr. Andrea Cavalli; Michele Vendruscolo
Angewandte Chemie 2011 Volume 123( Issue 41) pp:9794-9797
Publication Date(Web):
DOI:10.1002/ange.201101641
Co-reporter:Itzam De Gortari ; Guillem Portella ; Xavier Salvatella ; Vikram S. Bajaj ×; Patrick C. A. van der Wel ⊗; Jonathan R. Yates ; Matthew D. Segall ; Chris J. Pickard +; Mike C. Payne
Journal of the American Chemical Society 2010 Volume 132(Issue 17) pp:5993-6000
Publication Date(Web):April 13, 2010
DOI:10.1021/ja9062629
Since experimental measurements of NMR chemical shifts provide time and ensemble averaged values, we investigated how these effects should be included when chemical shifts are computed using density functional theory (DFT). We measured the chemical shifts of the N-formyl-l-methionyl-l-leucyl-l-phenylalanine-OMe (MLF) peptide in the solid state, and then used the X-ray structure to calculate the 13C chemical shifts using the gauge including projector augmented wave (GIPAW) method, which accounts for the periodic nature of the crystal structure, obtaining an overall accuracy of 4.2 ppm. In order to understand the origin of the difference between experimental and calculated chemical shifts, we carried out first-principles molecular dynamics simulations to characterize the molecular motion of the MLF peptide on the picosecond time scale. We found that 13C chemical shifts experience very rapid fluctuations of more than 20 ppm that are averaged out over less than 200 fs. Taking account of these fluctuations in the calculation of the chemical shifts resulted in an accuracy of 3.3 ppm. To investigate the effects of averaging over longer time scales we sampled the rotameric states populated by the MLF peptides in the solid state by performing a total of 5 μs classical molecular dynamics simulations. By averaging the chemical shifts over these rotameric states, we increased the accuracy of the chemical shift calculations to 3.0 ppm, with less than 1 ppm error in 10 out of 22 cases. These results suggests that better DFT-based predictions of chemical shifts of peptides and proteins will be achieved by developing improved computational strategies capable of taking into account the averaging process up to the millisecond time scale on which the chemical shift measurements report.
Co-reporter:Sebastian Pechmann and Michele Vendruscolo
Molecular BioSystems 2010 vol. 6(Issue 12) pp:2490-2497
Publication Date(Web):18 Oct 2010
DOI:10.1039/C005160H
Failure in maintaining protein solubility in vivo impairs protein homeostasis and results in protein misfolding and aggregation, which are often associated with severe neurodegenerative and systemic disorders that include Alzheimer's and Parkinson's diseases and type II diabetes. In this work we formulate a model of the competition between folding and aggregation, and derive a condition on the solubility of proteins in terms of the stability of their folded states, their aggregation propensities and their degradation rates. From our model, the bistability between folding and aggregation emerges as an intrinsic aspect of protein homeostasis. The analysis of the conditions that determine such a bistability provides a rationalization of the recently observed relationship between the cellular abundance and the aggregation propensity of proteins. We then discuss how the solubility condition that we derive can help rationalise the correlation that has been reported between evolutionary rates and expression levels or proteins, as well as in vivo protein solubility and expression level measurements, and recently elucidated trends of proteome evolution.
Co-reporter:Kai J. Kohlhoff ; Paul Robustelli ; Andrea Cavalli ; Xavier Salvatella
Journal of the American Chemical Society 2009 Volume 131(Issue 39) pp:13894-13895
Publication Date(Web):September 9, 2009
DOI:10.1021/ja903772t
We present a method, CamShift, for the rapid and accurate prediction of NMR chemical shifts from protein structures. The calculations performed by CamShift are based on an approximate expression of the chemical shifts in terms of polynomial functions of interatomic distances. Since these functions are very fast to compute and readily differentiable, the CamShift approach can be utilized in standard protein structure calculation protocols.
Co-reporter:Alfonso De Simone ; Andrea Cavalli ; Shang-Te Danny Hsu ; Wim Vranken
Journal of the American Chemical Society 2009 Volume 131(Issue 45) pp:16332-16333
Publication Date(Web):October 23, 2009
DOI:10.1021/ja904937a
We present a method for calculating accurate random coil chemical shift values of proteins. These values are obtained by analyzing the relationship between the amino acid sequences in flexible loop regions of native states and the corresponding experimentally measured chemical shifts. We estimate the errors in the random coil chemical shift scales to be 0.31 ppm for 13Cα, 0.37 ppm for 13Cβ, 0.31 ppm for 13CO, 0.68 ppm for 15N, 0.09 ppm for 1H, and 0.04 ppm for 1Hα.
Co-reporter:Jane R. Allison ; Peter Varnai ; Christopher M. Dobson
Journal of the American Chemical Society 2009 Volume 131(Issue 51) pp:18314-18326
Publication Date(Web):December 2, 2009
DOI:10.1021/ja904716h
Natively unfolded proteins present a challenge for structure determination because they populate highly heterogeneous ensembles of conformations. A useful source of structural information about these states is provided by paramagnetic relaxation enhancement measurements by nuclear magnetic resonance spectroscopy, from which long-range interatomic distances can be estimated. Here we describe a method for using such distances as restraints in molecular dynamics simulations to obtain a mapping of the free energy landscapes of natively unfolded proteins. We demonstrate the method in the case of α-synuclein and validate the results by a comparison with electron transfer measurements. Our findings indicate that our procedure provides an accurate estimate of the relative statistical weights of the different conformations populated by α-synuclein in its natively unfolded state.
Co-reporter:Paul Robustelli, Andrea Cavalli, Christopher M. Dobson, Michele Vendruscolo and Xavier Salvatella
The Journal of Physical Chemistry B 2009 Volume 113(Issue 22) pp:7890-7896
Publication Date(Web):May 8, 2009
DOI:10.1021/jp900780b
It has recently been shown that protein structures can be determined from nuclear magnetic resonance (NMR) chemical shifts using a molecular fragment replacement strategy. In these approaches, structural motifs are selected from existing protein structures on the basis of chemical shift and sequence homology and assembled to generate new structures. Here, we demonstrate that it is also possible to determine structures of proteins by directly incorporating experimental NMR chemical shifts as structural restraints in conformational searches, without the use of structural homology and molecular fragment replacement. In this approach, a protein is folded from an extended conformation to its native state using a simulated annealing procedure that minimizes an energy function that combines a standard force field with a term that penalizes the differences between experimental and calculated chemical shifts. We provide an initial demonstration of this procedure by determining the structure of two small proteins, with α and β folds, respectively.
Co-reporter:Emmanuel D. Levy;Sebastian Pechmann;Gian Gaetano Tartaglia
PNAS 2009 Volume 106 (Issue 25 ) pp:10159-10164
Publication Date(Web):2009-06-23
DOI:10.1073/pnas.0812414106
To maintain protein homeostasis, a variety of quality control mechanisms, such as the unfolded protein response and the heat
shock response, enable proteins to fold and to assemble into functional complexes while avoiding the formation of aberrant
and potentially harmful aggregates. We show here that a complementary contribution to the regulation of the interactions between
proteins is provided by the physicochemical properties of their amino acid sequences. The results of a systematic analysis
of the protein–protein complexes in the Protein Data Bank (PDB) show that interface regions are more prone to aggregate than
other surface regions, indicating that many of the interactions that promote the formation of functional complexes, including
hydrophobic and electrostatic forces, can potentially also cause abnormal intermolecular association. We also show, however,
that aggregation-prone interfaces are prevented from triggering uncontrolled assembly by being stabilized into their functional
conformations by disulfide bonds and salt bridges. These results indicate that functional and dysfunctional association of
proteins are promoted by similar forces but also that they are closely regulated by the presence of specific interactions
that stabilize native states.
Co-reporter:Gian Gaetano Tartaglia and Michele Vendruscolo
Chemical Society Reviews 2008 vol. 37(Issue 7) pp:1395-1401
Publication Date(Web):27 May 2008
DOI:10.1039/B706784B
Protein aggregation causes many devastating neurological and systemic diseases and represents a major problem in the preparation of recombinant proteins in biotechnology. Major advances in understanding the causes of this phenomenon have been made through the realisation that the analysis of the physico-chemical characteristics of the amino acids can provide accurate predictions about the rates of growth of the misfolded assemblies and the specific regions of the sequences that promote aggregation. More recently it has also been shown that the toxicity in vivo of protein aggregates can be predicted by estimating the propensity of polypeptide chains to form protofibrillar assemblies. In this tutorial review we describe the development of these predictions made through the Zyggregator method and the applications that have been explored so far.
Co-reporter:Andrea Cavalli;Xavier Salvatella;Christopher M. Dobson;
Proceedings of the National Academy of Sciences 2007 104(23) pp:9615-9620
Publication Date(Web):May 29, 2007
DOI:10.1073/pnas.0610313104
NMR spectroscopy plays a major role in the determination of the structures and dynamics of proteins and other biological macromolecules.
Chemical shifts are the most readily and accurately measurable NMR parameters, and they reflect with great specificity the
conformations of native and nonnative states of proteins. We show, using 11 examples of proteins representative of the major
structural classes and containing up to 123 residues, that it is possible to use chemical shifts as structural restraints
in combination with a conventional molecular mechanics force field to determine the conformations of proteins at a resolution
of 2 Å or better. This strategy should be widely applicable and, subject to further development, will enable quantitative
structural analysis to be carried out to address a range of complex biological problems not accessible to current structural
techniques.
Co-reporter:Joerg Gsponer;Harri Hopearuoho;Sara B.-M. Whittaker;Graham R. Spence;Geoffrey R. Moore;Emanuele Paci;Sheena E. Radford;
Proceedings of the National Academy of Sciences 2006 103(1) pp:99-104
Publication Date(Web):December 21, 2005
DOI:10.1073/pnas.0508667102
We present a detailed structural characterization of the intermediate state populated during the folding and unfolding of
the bacterial immunity protein Im7. We achieve this result by incorporating a variety of experimental data available for this
species in molecular dynamics simulations. First, we define the structure of the exchange-competent intermediate state of
Im7 by using equilibrium hydrogen-exchange protection factors. Second, we use this ensemble to predict Φ-values and compare
the results with the experimentally determined Φ-values of the kinetic refolding intermediate. Third, we predict chemical-shift
measurements and compare them with the measured chemical shifts of a mutational variant of Im7 for which the kinetic folding
intermediate is the most stable state populated at equilibrium. Remarkably, we found that the properties of the latter two
species are predicted with high accuracy from the exchange-competent intermediate that we determined, suggesting that these
three states are characterized by a similar architecture in which helices I, II, and IV are aligned in a native-like, but
reorganized, manner. Furthermore, the structural ensemble that we obtained enabled us to rationalize the results of tryptophan
fluorescence experiments in the WT protein and a series of mutational variants. The results show that the integration of diverse
sets of experimental data at relatively low structural resolution is a powerful approach that can provide insights into the
structural organization of this conformationally heterogeneous three-helix intermediate with unprecedented detail and highlight
the importance of both native and non-native interactions in stabilizing its structure.
Co-reporter:Kresten Lindorff-Larsen,
Robert B. Best,
Mark A. DePristo,
Christopher M. Dobson
and
Michele Vendruscolo
Nature 2005 433(7022) pp:128
Publication Date(Web):
DOI:10.1038/nature03199
Co-reporter:Xavier Salvatella;Christopher M. Dobson;Alan R. Fersht
PNAS 2005 102 (35 ) pp:12389-12394
Publication Date(Web):2005-08-30
DOI:10.1073/pnas.0408226102
The protein barnase folds from the denatured state into its native conformation via a high-energy intermediate. Using ΦI-values determined experimentally from single-point mutations as restraints in all-atom molecular dynamics simulations, we
have determined ensembles of structures corresponding to the transition states for the formation of the folding intermediate
and its conversion into the native state. We have also introduced a stringent validation of the approach used to calculate
such structures by predicting interaction ΦIJ-values determined experimentally from a series of double-mutant cycles. The ensembles that we have obtained illustrate the
heterogeneity in the nucleation-condensation process by which barnase folds. Obligatory steps of this process include the
sequential formation of two folding nuclei, which correspond to the two main hydrophobic cores of the protein. Nonobligatory
steps include the elongation of the strand β1 and the formation of the helix α2. The results confirm that the use of experimental
observables such as ΦI-values as restraints in molecular dynamics simulations is a powerful general strategy to characterize the relatively heterogeneous
structural ensembles that populate nonnative regions of the energy landscapes of proteins.
Co-reporter:Prajwal Ciryam, Rishika Kundra, Richard I. Morimoto, Christopher M. Dobson, Michele Vendruscolo
Trends in Pharmacological Sciences (February 2015) Volume 36(Issue 2) pp:72-77
Publication Date(Web):1 February 2015
DOI:10.1016/j.tips.2014.12.004
•Widespread protein aggregation is observed in neurodegenerative diseases.•Supersaturated proteins form a metastable sub-proteome prone to aggregation.•Specific biochemical pathways are enriched in supersaturated proteins.•Supersaturated pathways are associated with neurodegenerative diseases.The solubility of proteins is an essential requirement for their function. Nevertheless, these ubiquitous molecules can undergo aberrant aggregation when the protein homeostasis system becomes impaired. Here we ask: what are the driving forces for protein aggregation in the cellular environment? Emerging evidence suggests that this phenomenon arises at least in part because the native states of many proteins are inherently metastable when their cellular concentrations exceed their critical values. Such ‘supersaturated’ proteins, which form a ‘metastable subproteome’, are strongly driven towards aggregation, and are over-represented in specific biochemical pathways associated with neurodegenerative conditions. These observations suggest that effective therapeutic approaches designed to combat neurodegenerative diseases could be aimed at enhancing the ability of the cell to maintain the homeostasis of the metastable subproteome.
Co-reporter:Gian Gaetano Tartaglia, Andrea Cavalli, Michele Vendruscolo
Structure (February 2007) Volume 15(Issue 2) pp:139-143
Publication Date(Web):1 February 2007
DOI:10.1016/j.str.2006.12.007
Hydrogen exchange experiments provide detailed information about the local stability and the solvent accessibility of different regions of the structures of folded proteins, protein complexes, and amyloid fibrils. We introduce an approach to predict protection factors from hydrogen exchange in proteins based on the knowledge of their amino acid sequences without the inclusion of any additional structural information. These results suggest that the propensity of different regions of the structures of globular proteins to undergo local unfolding events can be predicted from their amino acid sequences with an accuracy of 80% or better.
Co-reporter:Gian Gaetano Tartaglia, Michele Vendruscolo
Journal of Molecular Biology (8 October 2010) Volume 402(Issue 5) pp:919-928
Publication Date(Web):8 October 2010
DOI:10.1016/j.jmb.2010.08.013
With the advent of proteomics, there is an increasing need of tools for predicting the properties of large numbers of proteins by using the information provided by their amino acid sequences, even in the absence of the knowledge of their structures. One of the most important types of predictions concerns whether proteins will fold or aggregate. Here, we study the competition between these two processes by analyzing the relationship between the folding and aggregation propensity profiles for the human and Escherichia coli proteomes. These profiles are calculated, respectively, using the CamFold method, which we introduce in this work, and the Zyggregator method. Our results indicate that the kinetic behavior of proteins is, to a large extent, determined by the interplay between regions of low folding and high aggregation propensities.
Co-reporter:Jörg Gsponer, John Christodoulou, Andrea Cavalli, Jennifer M. Bui, ... Michele Vendruscolo
Structure (7 May 2008) Volume 16(Issue 5) pp:736-746
Publication Date(Web):7 May 2008
DOI:10.1016/j.str.2008.02.017
We used nuclear magnetic resonance data to determine ensembles of conformations representing the structure and dynamics of calmodulin (CaM) in the calcium-bound state (Ca2+-CaM) and in the state bound to myosin light chain kinase (CaM-MLCK). These ensembles reveal that the Ca2+-CaM state includes a range of structures similar to those present when CaM is bound to MLCK. Detailed analysis of the ensembles demonstrates that correlated motions within the Ca2+-CaM state direct the structural fluctuations toward complex-like substates. This phenomenon enables initial ligation of MLCK at the C-terminal domain of CaM and induces a population shift among the substates accessible to the N-terminal domain, thus giving rise to the cooperativity associated with binding. Based on these results and the combination of modern free energy landscape theory with classical allostery models, we suggest that a coupled equilibrium shift mechanism controls the efficient binding of CaM to a wide range of ligands.
Co-reporter:Predrag Kukic, Hoi Tik Alvin Leung, Francesco Bemporad, Francesco A. Aprile, ... Michele Vendruscolo
Structure (7 April 2015) Volume 23(Issue 4) pp:745-753
Publication Date(Web):7 April 2015
DOI:10.1016/j.str.2014.12.020
•We identified three substates in the apo LFA-1 I-domain using RAM simulations•Independent RDC measurements were accurately reproduced using the three substates•The three substates correspond to the inactive, low, and intermediate affinity states•The three substates are associated with the precise regulation of the LFA-1 bindingLymphocyte function-associated antigen 1 (LFA-1) is an integrin that transmits information in two directions across the plasma membrane of leukocytes, in so-called outside-in and inside-out signaling mechanisms. To investigate the structural basis of these mechanisms, we studied the conformational space of the apo I-domain using replica-averaged metadynamics simulations in combination with nuclear magnetic resonance chemical shifts. We thus obtained a free energy landscape that reveals the existence of three conformational substates of this domain. The three substates include conformations similar to existing crystallographic structures of the low-affinity I-domain, the inactive I-domain with an allosteric antagonist inhibitor bound underneath α helix 7, and an intermediate affinity state of the I-domain. The multiple substates were validated with residual dipolar coupling measurements. These results suggest that the presence of three substates in the apo I-domain enables the precise regulation of the binding process that is essential for the physiological function of LFA-1.Download high-res image (151KB)Download full-size image
Co-reporter:Gian Gaetano Tartaglia, Amol P. Pawar, Silvia Campioni, Christopher M. Dobson, ... Michele Vendruscolo
Journal of Molecular Biology (4 July 2008) Volume 380(Issue 2) pp:425-436
Publication Date(Web):4 July 2008
DOI:10.1016/j.jmb.2008.05.013
We present a method for predicting the regions of the sequences of peptides and proteins that are most important in promoting their aggregation and amyloid formation. The method extends previous approaches by allowing such predictions to be carried out for conditions under which the molecules concerned can be folded or contain a significant degree of persistent structure. In order to achieve this result, the method uses only knowledge of the sequence of amino acids to estimate simultaneously both the propensity for folding and aggregation and the way in which these two types of propensity compete. We illustrate the approach by its application to a set of peptides and proteins both associated and not associated with disease. Our results show not only that the regions of a protein with a high intrinsic aggregation propensity can be identified in a robust manner but also that the structural context of such regions in the monomeric form is crucial for determining their actual role in the aggregation process.
Co-reporter:Pietro Sormanni, Francesco A. Aprile, Michele Vendruscolo
Journal of Molecular Biology (30 January 2015) Volume 427(Issue 2) pp:478-490
Publication Date(Web):30 January 2015
DOI:10.1016/j.jmb.2014.09.026
•Aggregation limits recombinant protein production and protein biologics development.•Antibodies are often poorly soluble at the concentrations needed for their delivery.•The CamSol method identifies protein mutants with enhanced solubility.•Tens of thousands of mutations can be screened in minutes on a laptop computer.Protein solubility is often an essential requirement in biotechnological and biomedical applications. Great advances in understanding the principles that determine this specific property of proteins have been made during the past decade, in particular concerning the physicochemical characteristics of their constituent amino acids. By exploiting these advances, we present the CamSol method for the rational design of protein variants with enhanced solubility. The method works by performing a rapid computational screening of tens of thousand of mutations to identify those with the greatest impact on the solubility of the target protein while maintaining its native state and biological activity. The application to a single-domain antibody that targets the Alzheimer's Aβ peptide demonstrates that the method predicts with great accuracy solubility changes upon mutation, thus offering a cost-effective strategy to help the production of soluble proteins for academic and industrial purposes.Download high-res image (141KB)Download full-size image
Co-reporter:Pietro Sormanni, Carlo Camilloni, Piero Fariselli, Michele Vendruscolo
Journal of Molecular Biology (27 February 2015) Volume 427(Issue 4) pp:982-996
Publication Date(Web):27 February 2015
DOI:10.1016/j.jmb.2014.12.007
•A sequence-based predictor is derived from NMR measurements of disordered proteins.•Order and disorder of proteins are predicted simultaneously.•Statistical weights are assigned to the ordered and disordered regions of proteins.•Structure and dynamics of proteins are characterized from their sequences.Extensive amounts of information about protein sequences are becoming available, as demonstrated by the over 79 million entries in the UniProt database. Yet, it is still challenging to obtain proteome-wide experimental information on the structural properties associated with these sequences. Fast computational predictors of secondary structure and of intrinsic disorder of proteins have been developed in order to bridge this gap. These two types of predictions, however, have remained largely separated, often preventing a clear characterization of the structure and dynamics of proteins. Here, we introduce a computational method to predict secondary-structure populations from amino acid sequences, which simultaneously characterizes structure and disorder in a unified statistical mechanics framework. To develop this method, called s2D, we exploited recent advances made in the analysis of NMR chemical shifts that provide quantitative information about the probability distributions of secondary-structure elements in disordered states. The results that we discuss show that the s2D method predicts secondary-structure populations with an average error of about 14%. A validation on three datasets of mostly disordered, mostly structured and partly structured proteins, respectively, shows that its performance is comparable to or better than that of existing predictors of intrinsic disorder and of secondary structure. These results indicate that it is possible to perform rapid and quantitative sequence-based characterizations of the structure and dynamics of proteins through the predictions of the statistical distributions of their ordered and disordered regions.Download high-res image (212KB)Download full-size image
Co-reporter:Michele Vendruscolo, Christopher M. Dobson
Current Biology (25 January 2011) Volume 21(Issue 2) pp:R68-R70
Publication Date(Web):25 January 2011
DOI:10.1016/j.cub.2010.11.062
The millisecond barrier has been broken in molecular dynamics simulations of proteins. Such simulations are increasingly revealing the inner workings of biological systems by generating atomic-level descriptions of their behaviour that make testable predictions about key molecular processes.
Co-reporter:Jin Chen, Hisashi Yagi, Pietro Sormanni, Michele Vendruscolo, ... Kunihiro Kuwajima
FEBS Letters (24 April 2012) Volume 586(Issue 8) pp:1120-1127
Publication Date(Web):24 April 2012
DOI:10.1016/j.febslet.2012.03.019
The chaperonin GroEL plays an essential role in promoting protein folding and in protecting against misfolding and aggregation in the cellular environment. In this study, we report that both GroEL and its isolated apical domain form amyloid-like fibrils under physiological conditions, and that the fibrillation of the apical domain is accelerated under acidic conditions. We also found, however, that despite its fibrillation propensity, the apical domain exhibits a pronounced inhibitory effect on the fibril growth of β2-microglobulin. Thus, the analysis of the behaviour of the apical domain reveals how aggregation and chaperone-mediated anti-aggregation processes can be closely related.Structured summary of protein interactionsgroEL and groEL bind by circular dichroism (View interaction)β2m and β2m bind by transmission electron microscopy (View interaction)β2m and β2m bind by fluorescence technology (View interaction)groEL and groEL bind by transmission electron microscopy (View interaction)groEL and groEL bind by nuclear magnetic resonance (View interaction)groEL and groEL bind by fluorescence technology (View interaction)Highlights► The fibrillogenic region of GroEL locates in its apical domain. ► Both GroEL and the apical domain can form amyloid-like fibrils in a physiological buffer. ► Isolated apical domain inhibits fibril-growth of human β2-microglobulin.
Co-reporter:Péter Várnai, Christopher M. Dobson, Michele Vendruscolo
Journal of Molecular Biology (21 March 2008) Volume 377(Issue 2) pp:575-588
Publication Date(Web):21 March 2008
DOI:10.1016/j.jmb.2008.01.012
Protein engineering techniques have emerged as powerful tools for characterizing transition states (TSs) for protein folding. Recently, the Ψ analysis, in which double-histidine mutations create the possibility of reversible crosslinking in the native state, has been proposed as an additional approach to the well-established Φ analysis. We present here a combination of these two procedures for defining the structure of the TS of ubiquitin, a small α/β protein that has been used extensively as a model system for both experimental and computational studies of the protein-folding process. We performed a series of molecular dynamics simulations in which Φ and Ψ values were used as ensemble-averaged structural restraints to determine an ensemble of structures representing the TS of ubiquitin. Although the available Ψ values for ubiquitin did not, by themselves, generate well-defined TS ensembles, the inclusion of the restricted set of zero or unity values, but not fractional ones, provided useful complementary information to the Φ analysis. Our results show that the TS of ubiquitin is formed by a relatively narrow ensemble of structures exhibiting an overall native-like topology in which the N-terminal and C-terminal regions are in close proximity.
Co-reporter:Katy E. Routledge, Gian Gaetano Tartaglia, Geoffrey W. Platt, Michele Vendruscolo, Sheena E. Radford
Journal of Molecular Biology (19 June 2009) Volume 389(Issue 4) pp:776-786
Publication Date(Web):19 June 2009
DOI:10.1016/j.jmb.2009.04.042
Despite much progress in understanding the folding and the aggregation processes of proteins, the rules defining their interplay have yet to be fully defined. This problem is of particular importance since many diseases are initiated by protein unfolding and hence the propensity to aggregate competes with intramolecular collapse and other folding events. Here, we describe the roles of intramolecular and intermolecular interactions in defining the length of the lag time and the apparent rate of elongation of the 100-residue protein human β2-microglobulin at pH 2.5, commencing from an acid-denatured state that lacks persistent structure but contains significant non-random hydrophobic interactions. Using a combination of site-directed mutagenesis, quantitative kinetic analysis and computational methods, we show that only a single region of about 10 residues in length, determines the rate of fibril formation, despite the fact that other regions exhibit a significant intrinsic propensity for aggregation. We rationalise these results by analysing the effect of incorporating the conformational properties of acid-unfolded β2-microglobulin and its variants at pH 2.5 as measured by NMR spectroscopy into the Zyggregator aggregation prediction algorithm. These results demonstrate that residual structure in the precursor state modulates the intrinsic propensity of the polypeptide chain to aggregate and that the algorithm developed here allows the key regions for aggregation to be more clearly identified and the rates of their self-association to be predicted. Given the common propensity of unfolded chains to form non-random intramolecular interactions as monomers and to self-assemble subsequently into amyloid fibrils, the approach developed should find widespread utility for the prediction of regions important in amyloid formation and their rates of self-assembly.
Co-reporter:Michele Vendruscolo
Journal of Molecular Biology (16 September 2011) Volume 412(Issue 2) pp:153-154
Publication Date(Web):16 September 2011
DOI:10.1016/j.jmb.2011.07.047
Co-reporter:Gian Gaetano Tartaglia, Christopher M. Dobson, F. Ulrich Hartl, Michele Vendruscolo
Journal of Molecular Biology (16 July 2010) Volume 400(Issue 3) pp:579-588
Publication Date(Web):16 July 2010
DOI:10.1016/j.jmb.2010.03.066
We describe a series of stringent relationships between abundance, solubility and chaperone usage of proteins. Based on these relationships, we show that the need of Escherichia coli proteins for the chaperonin GroEL can be predicted with 86% accuracy. Furthermore, from the observation that the abundance and solubility of proteins depend on the physicochemical properties of their amino acid sequences, we demonstrate that the requirement for GroEL can also be predicted directly from the sequences with 90% accuracy. These results indicate that the physicochemical properties of the amino acid sequences represent an essential component of the cellular quality control system that ensures the maintenance of protein homeostasis in living systems.
Co-reporter:Paul Robustelli, Andrea Cavalli, Michele Vendruscolo
Structure (12 December 2008) Volume 16(Issue 12) pp:1764-1769
Publication Date(Web):12 December 2008
DOI:10.1016/j.str.2008.10.016
Solid-state NMR spectroscopy does not require proteins to form crystalline or soluble samples and can thus be applied under a variety of conditions, including precipitates, gels, and microcrystals. It has recently been shown that NMR chemical shifts can be used to determine the structures of the native states of proteins in solution. By considering the cases of two proteins, GB1 and SH3, we provide an initial demonstration here that this type of approach can be extended to the use of solid-state NMR chemical shifts to obtain protein structures in the solid state without the need for measuring interatomic distances.
Co-reporter:Paul Robustelli, Kai Kohlhoff, Andrea Cavalli, Michele Vendruscolo
Structure (11 August 2010) Volume 18(Issue 8) pp:923-933
Publication Date(Web):11 August 2010
DOI:10.1016/j.str.2010.04.016
We introduce a procedure to determine the structures of proteins by incorporating NMR chemical shifts as structural restraints in molecular dynamics simulations. In this approach, the chemical shifts are expressed as differentiable functions of the atomic coordinates and used to compute forces to generate trajectories that lead to the reduction of the differences between experimental and calculated chemical shifts. We show that this strategy enables the folding of a set of proteins with representative topologies starting from partially denatured initial conformations without the use of additional experimental information. This method also enables the straightforward combination of chemical shifts with other standard NMR restraints, including those derived from NOE, J-coupling, and residual dipolar coupling measurements. We illustrate this aspect by calculating the structure of a transiently populated excited state conformation from chemical shift and residual dipolar coupling data measured by relaxation dispersion NMR experiments.Graphical AbstractDownload high-res image (330KB)Download full-size imageHighlights► Chemical shift restraints in molecular dynamics simulations ► Protein structure determination using chemical shift restraints ► Prediction of chemical shifts from protein structures ► Determination of the structures of protein intermediate states from NMR restraints
Co-reporter:Federico Agostini, Michele Vendruscolo, Gian Gaetano Tartaglia
Journal of Molecular Biology (10 August 2012) Volume 421(Issues 2–3) pp:237-241
Publication Date(Web):10 August 2012
DOI:10.1016/j.jmb.2011.12.005
In order to investigate the relationship between the thermodynamics and kinetics of protein aggregation, we compared the solubility of proteins with their aggregation rates. We found a significant correlation between these two quantities by considering a database of protein solubility values measured using an in vitro reconstituted translation system containing about 70% of Escherichia coli proteins. The existence of such correlation suggests that the thermodynamic stability of the native states of proteins relative to the aggregate states is closely linked with the kinetic barriers that separate them. In order to create the possibility of conducting computational studies at the proteome level to investigate further this concept, we developed a method of predicting the solubility of proteins based on their physicochemical properties.Download high-res image (120KB)Download full-size imageHighlights► Physicochemical principles of protein solubility. ► Accurate predictions of protein solubility. ► A link between thermodynamics and kinetics of protein aggregation.
Co-reporter:Gian Gaetano Tartaglia, Sebastian Pechmann, Christopher M. Dobson, Michele Vendruscolo
Journal of Molecular Biology (1 May 2009) Volume 388(Issue 2) pp:381-389
Publication Date(Web):1 May 2009
DOI:10.1016/j.jmb.2009.03.002
Each step in the process of gene expression, from the transcription of DNA into mRNA to the folding and posttranslational modification of proteins, is regulated by complex cellular mechanisms. At the same time, stringent conditions on the physicochemical properties of proteins, and hence on the nature of their amino acids, are imposed by the need to avoid aggregation at the concentrations required for optimal cellular function. A relationship is therefore expected to exist between mRNA expression levels and protein solubility in the cell. By investigating such a relationship, we formulate a method that enables the prediction of the maximal levels of mRNA expression in Escherichia coli with an accuracy of 83% and of the solubility of recombinant human proteins expressed in E. coli with an accuracy of 86%.
Co-reporter:Aditi N. Borkar, Pramodh Vallurupalli, Carlo Camilloni, Lewis E. Kay and Michele Vendruscolo
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 4) pp:NaN2804-2804
Publication Date(Web):2016/12/20
DOI:10.1039/C6CP08313G
RNA molecules in solution tend to undergo structural fluctuations of relatively large amplitude and to populate a range of different conformations some of which with low populations. It is still very challenging, however, to characterise the structures of these low populated states and to understand their functional roles. In the present study, we address this problem by using NMR residual dipolar couplings (RDCs) as structural restraints in replica-averaged metadynamics (RAM) simulations. By applying this approach to a 14-mer RNA hairpin containing the prototypical UUCG tetraloop motif, we show that it is possible to construct the free energy landscape of this RNA molecule. This free energy landscapes reveals the surprisingly rich dynamics of the UUCG tetraloop and identifies the multiple substates that exist in equilibrium owing to thermal fluctuations. The approach that we present is general and can be applied to the study of the free energy landscapes of other RNA or RNA-protein systems.
Co-reporter:Gian Gaetano Tartaglia and Michele Vendruscolo
Chemical Society Reviews 2008 - vol. 37(Issue 7) pp:NaN1401-1401
Publication Date(Web):2008/05/27
DOI:10.1039/B706784B
Protein aggregation causes many devastating neurological and systemic diseases and represents a major problem in the preparation of recombinant proteins in biotechnology. Major advances in understanding the causes of this phenomenon have been made through the realisation that the analysis of the physico-chemical characteristics of the amino acids can provide accurate predictions about the rates of growth of the misfolded assemblies and the specific regions of the sequences that promote aggregation. More recently it has also been shown that the toxicity in vivo of protein aggregates can be predicted by estimating the propensity of polypeptide chains to form protofibrillar assemblies. In this tutorial review we describe the development of these predictions made through the Zyggregator method and the applications that have been explored so far.

![3-[4-hydroxy-3-(5,5,8,8-tetramethyl-3-pentoxy-6,7-dihydronaphthalen-2-yl)phenyl]prop-2-enoic acid](http://img.cochemist.com/ccimg/847300/847239-17-2.png)
![3-[4-hydroxy-3-(5,5,8,8-tetramethyl-3-pentoxy-6,7-dihydronaphthalen-2-yl)phenyl]prop-2-enoic acid](http://img.cochemist.com/ccimg/847300/847239-17-2_b.png)
![5-{5-[2-chloro-4-(4,5-dihydro-1,3-oxazol-2-yl)phenoxy]pentyl}-3-methyl-1,2-oxazole](http://img.cochemist.com/ccimg/98100/98033-68-2.png)
![5-{5-[2-chloro-4-(4,5-dihydro-1,3-oxazol-2-yl)phenoxy]pentyl}-3-methyl-1,2-oxazole](http://img.cochemist.com/ccimg/98100/98033-68-2_b.png)











![Carbamic acid,N-[(1S)-2-[[(1S)-3-diazo-1-methyl-2-oxopropyl]amino]-2-oxo-1-(phenylmethyl)ethyl]-,phenylmethyl ester](http://img.cochemist.com/ccimg/71800/71732-53-1.png)
![Carbamic acid,N-[(1S)-2-[[(1S)-3-diazo-1-methyl-2-oxopropyl]amino]-2-oxo-1-(phenylmethyl)ethyl]-,phenylmethyl ester](http://img.cochemist.com/ccimg/71800/71732-53-1_b.png)


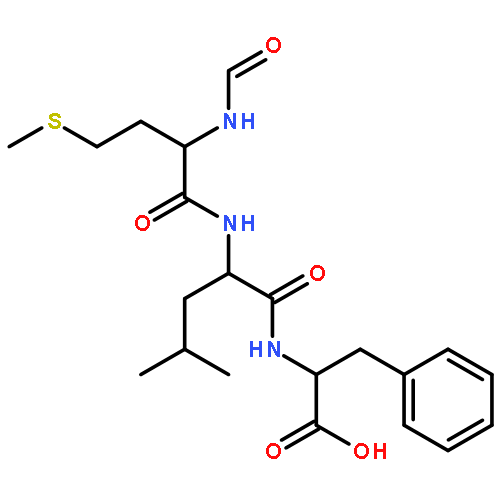
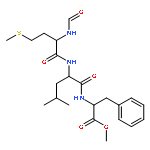
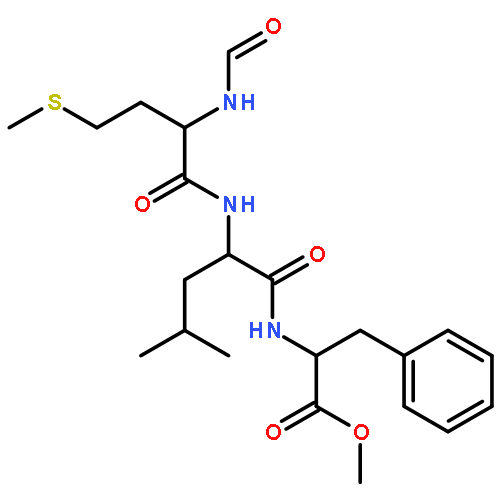
![4,6,10-Trioxa-5-phosphatetracosanoicacid, 2-amino-5-hydroxy-11-oxo-8-[(1-oxotetradecyl)oxy]-, 5-oxide, (2S,8R)-](http://img.cochemist.com/ccimg/64100/64023-32-1.png)
![4,6,10-Trioxa-5-phosphatetracosanoicacid, 2-amino-5-hydroxy-11-oxo-8-[(1-oxotetradecyl)oxy]-, 5-oxide, (2S,8R)-](http://img.cochemist.com/ccimg/64100/64023-32-1_b.png)
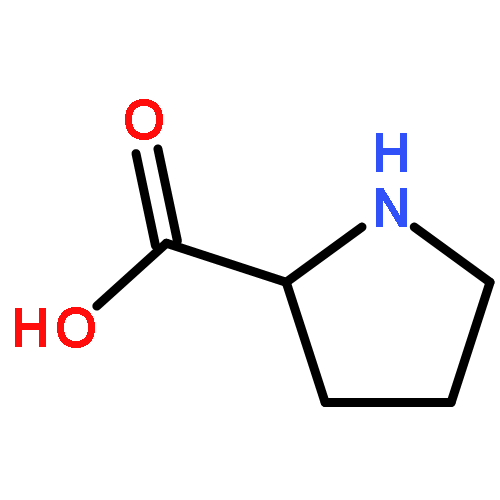


![Cholestane-7,24-diol,3-[[3-[(4-aminobutyl)amino]propyl]amino]-, 24-(hydrogen sulfate), (3b,5a,7a,24R)-](http://img.cochemist.com/ccimg/148800/148717-90-2.png)
![Cholestane-7,24-diol,3-[[3-[(4-aminobutyl)amino]propyl]amino]-, 24-(hydrogen sulfate), (3b,5a,7a,24R)-](http://img.cochemist.com/ccimg/148800/148717-90-2_b.png)
![Benzoic acid,4-(7,8,9,10-tetrahydro-5,7,7,10,10-pentamethyl-5H-benzo[e]naphtho[2,3-b][1,4]diazepin-13-yl)-](http://img.cochemist.com/ccimg/155900/155877-83-1.png)
![Benzoic acid,4-(7,8,9,10-tetrahydro-5,7,7,10,10-pentamethyl-5H-benzo[e]naphtho[2,3-b][1,4]diazepin-13-yl)-](http://img.cochemist.com/ccimg/155900/155877-83-1_b.png)






![Benzoic acid,4-[(1E)-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/71500/71441-28-6.png)
![Benzoic acid,4-[(1E)-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/71500/71441-28-6_b.png)
![4-[2-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-1,3-dioxolan-2-yl]benzoic acid](http://img.cochemist.com/ccimg/146700/146670-40-8.png)
![4-[2-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-1,3-dioxolan-2-yl]benzoic acid](http://img.cochemist.com/ccimg/146700/146670-40-8_b.png)
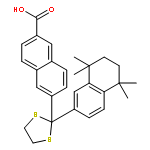
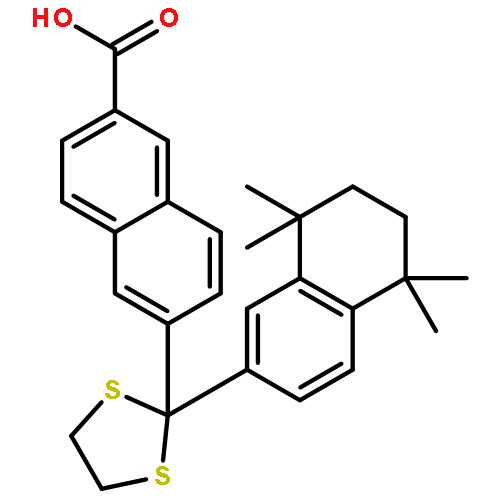


![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1_b.png)



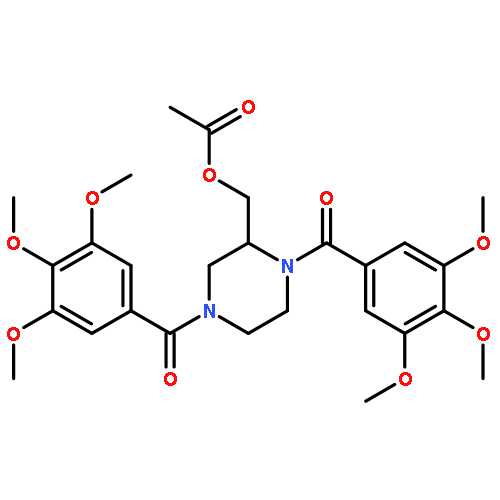
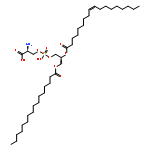
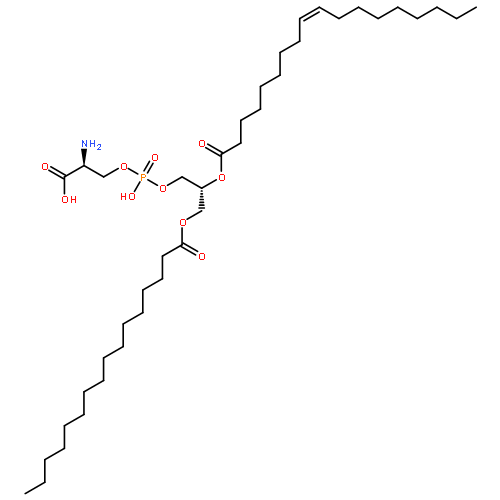


![4-[6-(1-adamantyl)-7-hydroxy-2-naphthyl]benzoic acid](http://img.cochemist.com/ccimg/107500/107430-66-0.png)
![4-[6-(1-adamantyl)-7-hydroxy-2-naphthyl]benzoic acid](http://img.cochemist.com/ccimg/107500/107430-66-0_b.png)
![Benzoic acid,4-[(1E)-3-[3,5-bis(1,1-dimethylethyl)phenyl]-3-oxo-1-propen-1-yl]-](http://img.cochemist.com/ccimg/110400/110368-33-7.png)
![Benzoic acid,4-[(1E)-3-[3,5-bis(1,1-dimethylethyl)phenyl]-3-oxo-1-propen-1-yl]-](http://img.cochemist.com/ccimg/110400/110368-33-7_b.png)


![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)
![Sodium;(2s)-2-azaniumyl-3-[[(2r)-2,3-di(hexadecanoyloxy)propoxy]-oxidophosphoryl]oxypropanoate](http://img.cochemist.com/ccimg/145900/145849-32-7.png)
![Sodium;(2s)-2-azaniumyl-3-[[(2r)-2,3-di(hexadecanoyloxy)propoxy]-oxidophosphoryl]oxypropanoate](http://img.cochemist.com/ccimg/145900/145849-32-7_b.png)



















![2-(4-{3-[2-(TRIFLUOROMETHYL)-10H-PHENOTHIAZIN-10-YL]PROPYL}-1-PIP<WBR />ERAZINYL)ETHYL HEPTANOATE](http://img.cochemist.com/ccimg/215100/215030-90-3.png)
![2-(4-{3-[2-(TRIFLUOROMETHYL)-10H-PHENOTHIAZIN-10-YL]PROPYL}-1-PIP<WBR />ERAZINYL)ETHYL HEPTANOATE](http://img.cochemist.com/ccimg/215100/215030-90-3_b.png)