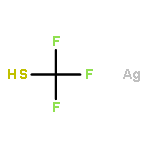
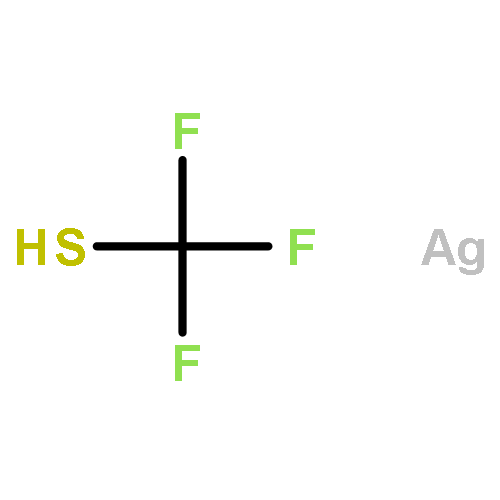
Co-reporter:Jiean Chen, Pengfei Yuan, Leming Wang, and Yong Huang
Journal of the American Chemical Society May 24, 2017 Volume 139(Issue 20) pp:7045-7045
Publication Date(Web):May 5, 2017
DOI:10.1021/jacs.7b02889
Remote asymmetric protonation is a longstanding challenge due to the small size of protons. Reactions involving electron-deficient olefins pose a further difficulty due to the electrophilic nature of these substrates. We report a shuttling system that delivers a proton in a highly enantioselective manner to the β-carbon of enals using a chiral N-heterocyclic carbene (NHC) catalyst. Choices of a Brønsted base shuttle and a Brønsted acid cocatalyst are critical for highly stereoselective β-protonation of the homoenolate intermediate and regeneration of the NHC catalyst results in functionalization of the carbonyl group. Thioesters with a β-chiral center were prepared in a redox-neutral transformation with an excellent yield and ee.
Co-reporter:Leifeng Wang;Fengjin Wu;Dr. Jiean Chen; Dr. David A. Nicewicz; Dr. Yong Huang
Angewandte Chemie 2017 Volume 129(Issue 24) pp:7000-7004
Publication Date(Web):2017/06/06
DOI:10.1002/ange.201702940
AbstractWe report a formal [4+2] cycloaddition reaction of styrenes under visible-light catalysis. Two styrene molecules with different electronic or steric properties were found to react with each other in good yield and excellent chemo- and regioselectivity. This reaction provides direct access to polysubstituted tetralin scaffolds from readily available styrenes. Sophisticated tricyclic and tetracyclic tetralin analogues were prepared in high yield and up to 20/1 diasteroselectivity from cyclic substrates.
Co-reporter:Leifeng Wang;Fengjin Wu;Dr. Jiean Chen; Dr. David A. Nicewicz; Dr. Yong Huang
Angewandte Chemie International Edition 2017 Volume 56(Issue 24) pp:6896-6900
Publication Date(Web):2017/06/06
DOI:10.1002/anie.201702940
AbstractWe report a formal [4+2] cycloaddition reaction of styrenes under visible-light catalysis. Two styrene molecules with different electronic or steric properties were found to react with each other in good yield and excellent chemo- and regioselectivity. This reaction provides direct access to polysubstituted tetralin scaffolds from readily available styrenes. Sophisticated tricyclic and tetracyclic tetralin analogues were prepared in high yield and up to 20/1 diasteroselectivity from cyclic substrates.
Co-reporter:Zhiqi He and Yong Huang
ACS Catalysis 2016 Volume 6(Issue 11) pp:7814
Publication Date(Web):October 4, 2016
DOI:10.1021/acscatal.6b02477
Direct homologation of aromatic amides with internal alkynes has been accomplished via a nickel-catalyzed sequential C–H activation reaction. The use of a rigid chelating group and a strong aprotic polar solvent successfully divert the classical [4 + 2] annulation to the [2 + 2 + 2] homologation pathway. This transformation is promoted by a simple nickel catalyst without the need of stoichiometric metal oxidants. Mechanistic studies support an unusual substrate-assisted ligand exchange process. NMR and X-ray data suggest a [5,5] Ni-bridged metallacycle as the catalyst resting state. Substrate assisted directing group swap plays an important role for the subsequent meta-C-H insertion. In contrast, [4 + 2] annulation can be accomplished using a bulky, electron-rich phosphine ligand, which favors rapid reductive C–N elimination.Keywords: C−H activation; dehydrogenative homologation; directing group; nickel catalysis; phosphine
Co-reporter:Jiean Chen
Science China Chemistry 2016 Volume 59( Issue 3) pp:251-254
Publication Date(Web):2016 March
DOI:10.1007/s11426-015-5514-7
Co-reporter:Jiean Chen, Sixuan Meng, Leming Wang, Hongmei Tang and Yong Huang
Chemical Science 2015 vol. 6(Issue 7) pp:4184-4189
Publication Date(Web):23 Apr 2015
DOI:10.1039/C5SC00878F
We report the first asymmetric sulfa-Michael addition (SMA) reactions using a chiral N-heterocyclic carbene (NHC) as a non-covalent organocatalyst. We demonstrate that a triazolium salt derived NHC functions as an excellent Brønsted base to promote enantioselective carbon–sulfur bond formation. The reaction is applicable to a wide range of thiols and electrophilic olefins. Notably, quaternary chiral centers bearing both an S atom and a CF3 group were synthesized with excellent asymmetric control. Mechanistic studies suggest that the facial discrimination is likely to be guided by non-covalent interactions: hydrogen bonding and π–π stacking.
Co-reporter:Qian Shao and Yong Huang
Chemical Communications 2015 vol. 51(Issue 30) pp:6584-6586
Publication Date(Web):06 Mar 2015
DOI:10.1039/C5CC01407G
We have developed a practical method to synthesize fluorostyrene compounds. A mild and regioselective mono-fluorination reaction occurred smoothly for various di- and trisubstituted styrenes in the presence of RuCl3 and N-fluorobenzenesulfonimide (NFSI). A tandem alkyne hydroarylation–olefin fluorination reaction was also developed using an Au catalyst.
Co-reporter:Leming Wang;Dr. Jiean Chen;Dr. Yong Huang
Angewandte Chemie International Edition 2015 Volume 54( Issue 51) pp:15414-15418
Publication Date(Web):
DOI:10.1002/anie.201508371
Abstract
The aza-Michael addition reaction is a vital transformation for the synthesis of functionalized chiral amines. Despite intensive research, enantioselective aza-Michael reactions with alkyl amines as the nitrogen donor have not been successful. We report the use of chiral N-heterocyclic carbenes (NHCs) as noncovalent organocatalysts to promote a highly selective aza-Michael reaction between primary alkyl amines and β-trifluoromethyl β-aryl nitroolefins. In contrast to classical conjugate-addition reactions, a strategy of HOMO-raising activation was used. Chiral trifluoromethylated amines were synthesized in high yield (up to 99 %) with excellent enantioselectivity (up to 98 % ee).
Co-reporter:Leming Wang;Dr. Jiean Chen;Dr. Yong Huang
Angewandte Chemie 2015 Volume 127( Issue 51) pp:15634-15638
Publication Date(Web):
DOI:10.1002/ange.201508371
Abstract
The aza-Michael addition reaction is a vital transformation for the synthesis of functionalized chiral amines. Despite intensive research, enantioselective aza-Michael reactions with alkyl amines as the nitrogen donor have not been successful. We report the use of chiral N-heterocyclic carbenes (NHCs) as noncovalent organocatalysts to promote a highly selective aza-Michael reaction between primary alkyl amines and β-trifluoromethyl β-aryl nitroolefins. In contrast to classical conjugate-addition reactions, a strategy of HOMO-raising activation was used. Chiral trifluoromethylated amines were synthesized in high yield (up to 99 %) with excellent enantioselectivity (up to 98 % ee).
Co-reporter:Hu Chen, Qian Wang, Yong Huang
Tetrahedron 2015 Volume 71(Issue 22) pp:3632-3636
Publication Date(Web):3 June 2015
DOI:10.1016/j.tet.2014.11.071
A convenient and efficient copper-catalyzed aerobic cascade reaction has been developed for the synthesis of the pharmacologically relevant isoindolin-1-ylidene scaffold. We discovered that various ortho-formyl cinnamates could react smoothly with different amines in the presence of a commercially available copper catalyst under mild aerobic conditions. Isoindolin-1-ylidene derivatives were assembled in one pot in moderate to good yields. This method features amine annulation and double dehydrogenation, representing high atomic efficiency. Its product could be further converted to the privileged isoindolinone pharmacophore.
Co-reporter:Zhaofeng Wang ; Li Li
Journal of the American Chemical Society 2014 Volume 136(Issue 35) pp:12233-12236
Publication Date(Web):August 18, 2014
DOI:10.1021/ja506352b
We describe a direct synthesis of various ynones from readily available aldehydes and hypervalent alkynyl iodides. In this method, a gold catalyst and a secondary amine work synergistically to produce the trisubstituted allenyl aldehyde, which can be converted to the desired ynone through an in situ C–C bond oxidative cleavage using molecular oxygen.
Co-reporter:Zhenxing Zhang, Hao Jiang, and Yong Huang
Organic Letters 2014 Volume 16(Issue 22) pp:5976-5979
Publication Date(Web):November 7, 2014
DOI:10.1021/ol502998n
The first Ru-catalyzed redox-neutral C–H activation reaction via N–N bond cleavage is reported. Pyrazolidin-3-one is demonstrated as an internally oxidative directing group that enables C–H annulation reactions with a broad scope of alkynes, including previously incompetent terminal alkynes. Pharmacologically privileged 3-(1H-indol-1-yl)propanamides were synthesized in high yields.
Co-reporter:Weiyu Yin;Zhaofeng Wang
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 14-15) pp:2998-3006
Publication Date(Web):
DOI:10.1002/adsc.201400362
Co-reporter:Yan Fang, Chengming Wang, Shengqin Su, Haizhu Yu and Yong Huang
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 7) pp:1061-1071
Publication Date(Web):03 Dec 2013
DOI:10.1039/C3OB42088D
We described two orthogonal heterocycle syntheses, where an arene bearing both an alkyne and a triazene functionality underwent two distinct cyclization pathways mediated by different transition metals. Starting from the same substrates, a synthesis of 2H-indazole was accomplished by a Cu(II) salt promoted oxidative cyclization, while 2-substituted indoles could be accessed via a Ag(I) salt mediated N–N bond cleavage. This method represents the first synthesis of indoles from alkynyl triazenes. Computational analysis was performed for both reaction pathways, supporting a Lewis acid role for Cu and a π-acid catalysis for Ag.
Co-reporter:Tao Cheng, Weiyu Yin, Yi Zhang, Yingnan Zhang and Yong Huang
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 9) pp:1405-1411
Publication Date(Web):20 Dec 2013
DOI:10.1039/C3OB42196A
A general palladium catalyzed acetoxylation of benzylic C–H bonds has been developed. Picolinamides serve as an excellent directing group for the C–H activation of benzylic methyls. A wide range of 2-amino benzyl alcohol analogues were synthesized in good yields. The products demonstrated broad synthetic utilities toward various benzo-fused heterocycles. Mechanistic studies revealed the key rate-limiting C–H insertion step, which could be affected by the substitution pattern of the parent arene.
Co-reporter:Yi Zhang, Qian Wang, Huidong Yu and Yong Huang
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 44) pp:8844-8850
Publication Date(Web):29 Jul 2014
DOI:10.1039/C4OB01312C
We describe a straightforward protocol for a smooth dehydrogenative annulation reaction between various arenes and terminal alkynes using a catalytic amount of CuBr2 and molecular oxygen. 3-Methyleneisoindoline derivatives are prepared in high yields.
Co-reporter:Huan Sun, Chengming Wang, Yun-Fang Yang, Ping Chen, Yun-Dong Wu, Xinhao Zhang, and Yong Huang
The Journal of Organic Chemistry 2014 Volume 79(Issue 24) pp:11863-11872
Publication Date(Web):May 13, 2014
DOI:10.1021/jo500807d
Indole-containing polyaromatic scaffolds are widely found in natural products, pharmaceutical agents, and π-conjugated functional materials. Often, the synthesis of these highly valuable molecules requires a multistep sequence. Therefore, a simple, one-step protocol to access libraries of polyaromatic indole scaffolds is highly desirable. Herein we describe the direct synthesis of polysubstituted indolo[2,1-a]isoquinoline analogues via a double C–H annulation cascade using triazene as an internally cleavable directing group. Evidence from HRMS and theoretical calculations suggests that an unprecedented 1,2-alkyl migration might be responsible for the in situ cleavage of the directing group. Both kinetic isotope effects and DFT calculations suggested that the alkyne insertion step is rate-limiting for the second C,N annulation reaction.
Co-reporter:Chaosheng Luo
Journal of the American Chemical Society 2013 Volume 135(Issue 22) pp:8193-8196
Publication Date(Web):May 16, 2013
DOI:10.1021/ja4040945
Tetrahydroquinolines containing two quaternary stereogenic centers were synthesized with excellent ee and dr via a four-component cyclization reaction catalyzed by a chiral phosphoric acid. High chemoselectivity was achieved by differentiating anilines with similar reactivities to yield diverse “hybrid” products. The chirality of the quaternary C4 atom of the 4-aminotetrahydroquinoline products was found to undergo highly stereoselective inversion, enabling facile functionalization using a wide range of nucleophiles (C, O, N, and S).
Co-reporter:Chengming Wang and Yong Huang
Organic Letters 2013 Volume 15(Issue 20) pp:5294-5297
Publication Date(Web):October 7, 2013
DOI:10.1021/ol402523x
A general protocol for the synthesis of N-alkyl indoles has been developed via a redox neutral C–H activation strategy using a traceless nitroso directing group. A broad scope of substituted N-alkyl indoles has been prepared in good to excellent yields using a very simple Rh catalyst system in the absence of an external oxidant or any other additive. Good to excellent regioselectivity has been achieved for asymmetrically disubstituted acetylenes.
Co-reporter:Weiyu Yin, Chengming Wang, and Yong Huang
Organic Letters 2013 Volume 15(Issue 8) pp:1850-1853
Publication Date(Web):April 5, 2013
DOI:10.1021/ol400459y
A mild, aerobic, catalytic process for obtaining nitriles directly from alcohols and aqueous ammonia is described. The reaction proceeds via a dehydrogenation cascade mediated by catalytic CuI, bpy, and TEMPO in the presence of O2. The substrate scope is broad including various functionalized aromatic and aliphatic alcohols. This protocol enabled the one-pot synthesis of various biaryl heterocycles directly from commercially available alcohols.
Co-reporter:Qian Shao, Jiean Chen, Meihua Tu, David W. Piotrowski and Yong Huang
Chemical Communications 2013 vol. 49(Issue 94) pp:11098-11100
Publication Date(Web):09 Oct 2013
DOI:10.1039/C3CC46757K
An enantioselective organocatalytic process for the one-step synthesis of poly-substituted 1,2,4-triazolines is reported. The heterocycle formation is believed to go through a step-wise mechanism of nucleophilic addition of an azlactone to an azodicarboxylate in the presence of an organic base catalyst, followed by a TMSCHN2 mediated heterocyclization. Both theoretical calculations and experimental evidence suggest the pre-organization of the transition state for the chirality determining step via a unique 7-membered intramolecular hydrogen bonding.
Co-reporter:Tao Cheng, Sixuan Meng, and Yong Huang
Organic Letters 2013 Volume 15(Issue 8) pp:1958-1961
Publication Date(Web):April 8, 2013
DOI:10.1021/ol4006129
Highly functionalized pyrrolidine and piperidine analogues, with up to three stereogenic centers, were synthesized in good yield (50–95%), excellent dr (single isomer), and high ee (>90%) using a Cinchona alkaloid-derived carbamate organocatalyst. High stereoselective synergy was achieved by combining a reversible aza-Henry reaction with a dynamic kinetic resolution (DKR)-driven aza-Michael cyclization. Whereas both reactions proceed with moderate enantioselectivities (50–60% for each step), high enantioselectivities are obtained for the overall products devoid of dr sacrifice.
Co-reporter:Qi Zhang, Hai-Zhu Yu, Yi-Tong Li, Lei Liu, Yong Huang and Yao Fu
Dalton Transactions 2013 vol. 42(Issue 12) pp:4175-4184
Publication Date(Web):06 Feb 2013
DOI:10.1039/C3DT31898B
A systematic theoretical study on the Rh-catalyzed oxidative Heck-coupling of phenol carbamates with alkenes is carried out. Two possible mechanisms (i.e. arene activation-first and alkene activation-first mechanisms) are examined. As to the C–H activation step, four mechanisms including oxidative addition, electrophilic substitution, concerted metallation-deprotonation (CMD), and σ-bond metathesis are evaluated. The calculation results indicate that the arene activation-first mechanism is more favorable for the overall catalytic cycle. This mechanism involves three steps: arene C–H activation at the position ortho to the carbamate directing group affording a six-membered rhodiacycle intermediate, insertion of the alkene double bond into the Rh(III)–aryl bond, and a final β-H elimination step to release the product and re-generate the catalyst. The rate determining step of the overall catalytic cycle is the arene C–H activation step, which is found to proceed through the acetate-assisted CMD mechanism.
Co-reporter:Chengming Wang;Huan Sun;Yan Fang ;Dr. Yong Huang
Angewandte Chemie International Edition 2013 Volume 52( Issue 22) pp:
Publication Date(Web):
DOI:10.1002/anie.201303274
Co-reporter:Zhaofeng Wang;Xijian Li ;Dr. Yong Huang
Angewandte Chemie 2013 Volume 125( Issue 52) pp:14469-14473
Publication Date(Web):
DOI:10.1002/ange.201308835
Abstract
Carbonyl-substituted allenes are highly important synthetic intermediates for a number of heterocycles and strained-ring systems. However, chemistry of allenyl aldehydes has not been explored as extensively as their ketone, ester, or amide analogues because of a lack of general synthetic methods. Described herein is the first direct α-vinylidenation of aldehydes and an α-vinylidenation/γ-functionalization cascade to access tri- and tetrasubstituted allenyl aldehydes using a combination of a gold catalyst and an secondary amine. The reactive enamine intermediate of an aldehyde reacts with the gold-activated hypervalent silylethynyl benziodoxolone to selectively generate the corresponding trisubstituted allenyl aldehyde. The allenyl aldehyde can further react with another equivalent of the alkynylation reagent or other electrophiles to afford tetrasubstituted allenes bearing an aldehyde group, an acetylene, and a halogen functionality. This method enables rapid access to polysubstituted furans from aldehydes.
Co-reporter:Zhaofeng Wang;Xijian Li ;Dr. Yong Huang
Angewandte Chemie International Edition 2013 Volume 52( Issue 52) pp:14219-14223
Publication Date(Web):
DOI:10.1002/anie.201308835
Abstract
Carbonyl-substituted allenes are highly important synthetic intermediates for a number of heterocycles and strained-ring systems. However, chemistry of allenyl aldehydes has not been explored as extensively as their ketone, ester, or amide analogues because of a lack of general synthetic methods. Described herein is the first direct α-vinylidenation of aldehydes and an α-vinylidenation/γ-functionalization cascade to access tri- and tetrasubstituted allenyl aldehydes using a combination of a gold catalyst and an secondary amine. The reactive enamine intermediate of an aldehyde reacts with the gold-activated hypervalent silylethynyl benziodoxolone to selectively generate the corresponding trisubstituted allenyl aldehyde. The allenyl aldehyde can further react with another equivalent of the alkynylation reagent or other electrophiles to afford tetrasubstituted allenes bearing an aldehyde group, an acetylene, and a halogen functionality. This method enables rapid access to polysubstituted furans from aldehydes.
Co-reporter:Chengming Wang;Huan Sun;Yan Fang ;Dr. Yong Huang
Angewandte Chemie International Edition 2013 Volume 52( Issue 22) pp:5795-5798
Publication Date(Web):
DOI:10.1002/anie.201301742
Co-reporter:Zhaofeng Wang;Xijian Li ;Dr. Yong Huang
Angewandte Chemie 2013 Volume 125( Issue 52) pp:
Publication Date(Web):
DOI:10.1002/ange.201310080
Co-reporter:Chengming Wang;Huan Sun;Yan Fang ;Dr. Yong Huang
Angewandte Chemie 2013 Volume 125( Issue 22) pp:5907-5910
Publication Date(Web):
DOI:10.1002/ange.201301742
Co-reporter:Chengming Wang;Huan Sun;Yan Fang ;Dr. Yong Huang
Angewandte Chemie 2013 Volume 125( Issue 22) pp:
Publication Date(Web):
DOI:10.1002/ange.201303274
Co-reporter:Hu Chen, Zhaofeng Wang, Yingnan Zhang, and Yong Huang
The Journal of Organic Chemistry 2013 Volume 78(Issue 7) pp:3503-3509
Publication Date(Web):March 15, 2013
DOI:10.1021/jo400215e
A triple cascade was developed using a simple copper catalyst to trans-selectively access bicyclic isoxazolidines in a one-pot synthesis. This strategy features the in situ generation of nitrones and subsequent trapping by [3 + 2] cycloaddition. In this method, copper serves three catalytic functions: as a Lewis acid for the ene reaction, as an organometallic for aerobic oxidation, and as a Lewis acid for an endo-selective [3 + 2] cycloaddition. The successful merging of aerobic oxidation and Lewis acid catalysis demonstrated efficient cascade synergy.
Co-reporter:Zhaofeng Wang;Xijian Li ;Dr. Yong Huang
Angewandte Chemie International Edition 2013 Volume 52( Issue 52) pp:
Publication Date(Web):
DOI:10.1002/anie.201310080
Co-reporter:Guiyong Wu, Weiyu Yin, Hong C. Shen and Yong Huang
Green Chemistry 2012 vol. 14(Issue 3) pp:580-585
Publication Date(Web):12 Jan 2012
DOI:10.1039/C2GC16457D
To access useful heterocycles in medicinal chemistry such as pyridazinones, dihydropyrimidinones, and dihydropyrimidinthiones, a “green” mild and highly efficient one-pot triple cascade was developed involving a Claisen–decarboxylation, electrophilic reaction, and subsequent heterocyclization. In addition, indazoles and benzofurans could also be constructed via a double cascade. To develop the cascade process, a direct Claisen–decarboxylation reaction was firstly optimized. This reaction can then couple with electrophilic reactions including alkylation, Michael addition or aldol reaction to enable the preparation of various aryl ketones in a one-pot fashion.
Co-reporter:Weiming Gao, Zhiqi He, Yong Qian, Jing Zhao and Yong Huang
Chemical Science 2012 vol. 3(Issue 3) pp:883-886
Publication Date(Web):07 Nov 2011
DOI:10.1039/C1SC00661D
We describe a general dehydrogenation procedure to form α,β-unsaturated aldehydes, ketones, esters and azobenzenes under very mild conditions, requiring catalytic commercial Pd(OAc)2, a catalytic weak inorganic base and air as the sole oxidant. In the presence of a diazafluorenone ligand, this process converts aliphatic aldehydes to α,β-unsaturated aldehydes in an open-flask fashion at ambient pressure and temperature. A broad spectrum of substrates, including aldehydes, ketones, esters, alcohols and hydrazines, were conveniently dehydrogenated under a relatively uniformed protocol. A mechanism involving β-elimination-driven enolization equilibrium shift was proposed.
Co-reporter:Chengming Wang;Hu Chen;Zhaofeng Wang;Jiean Chen ;Dr. Yong Huang
Angewandte Chemie 2012 Volume 124( Issue 29) pp:7354-7357
Publication Date(Web):
DOI:10.1002/ange.201203230
Co-reporter:Chengming Wang;Hu Chen;Zhaofeng Wang;Jiean Chen ;Dr. Yong Huang
Angewandte Chemie International Edition 2012 Volume 51( Issue 29) pp:
Publication Date(Web):
DOI:10.1002/anie.201204565
Co-reporter:Chengming Wang;Hu Chen;Zhaofeng Wang;Jiean Chen ;Dr. Yong Huang
Angewandte Chemie International Edition 2012 Volume 51( Issue 29) pp:7242-7245
Publication Date(Web):
DOI:10.1002/anie.201203230
Co-reporter:Chengming Wang;Hu Chen;Zhaofeng Wang;Jiean Chen ;Dr. Yong Huang
Angewandte Chemie 2012 Volume 124( Issue 29) pp:
Publication Date(Web):
DOI:10.1002/ange.201204565
Co-reporter:Yi Zhang, Qian Wang, Huidong Yu and Yong Huang
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 44) pp:NaN8850-8850
Publication Date(Web):2014/07/29
DOI:10.1039/C4OB01312C
We describe a straightforward protocol for a smooth dehydrogenative annulation reaction between various arenes and terminal alkynes using a catalytic amount of CuBr2 and molecular oxygen. 3-Methyleneisoindoline derivatives are prepared in high yields.
Co-reporter:Qian Shao and Yong Huang
Chemical Communications 2015 - vol. 51(Issue 30) pp:NaN6586-6586
Publication Date(Web):2015/03/06
DOI:10.1039/C5CC01407G
We have developed a practical method to synthesize fluorostyrene compounds. A mild and regioselective mono-fluorination reaction occurred smoothly for various di- and trisubstituted styrenes in the presence of RuCl3 and N-fluorobenzenesulfonimide (NFSI). A tandem alkyne hydroarylation–olefin fluorination reaction was also developed using an Au catalyst.
Co-reporter:Weiming Gao, Zhiqi He, Yong Qian, Jing Zhao and Yong Huang
Chemical Science (2010-Present) 2012 - vol. 3(Issue 3) pp:NaN886-886
Publication Date(Web):2011/11/07
DOI:10.1039/C1SC00661D
We describe a general dehydrogenation procedure to form α,β-unsaturated aldehydes, ketones, esters and azobenzenes under very mild conditions, requiring catalytic commercial Pd(OAc)2, a catalytic weak inorganic base and air as the sole oxidant. In the presence of a diazafluorenone ligand, this process converts aliphatic aldehydes to α,β-unsaturated aldehydes in an open-flask fashion at ambient pressure and temperature. A broad spectrum of substrates, including aldehydes, ketones, esters, alcohols and hydrazines, were conveniently dehydrogenated under a relatively uniformed protocol. A mechanism involving β-elimination-driven enolization equilibrium shift was proposed.
Co-reporter:Yan Fang, Chengming Wang, Shengqin Su, Haizhu Yu and Yong Huang
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 7) pp:NaN1071-1071
Publication Date(Web):2013/12/03
DOI:10.1039/C3OB42088D
We described two orthogonal heterocycle syntheses, where an arene bearing both an alkyne and a triazene functionality underwent two distinct cyclization pathways mediated by different transition metals. Starting from the same substrates, a synthesis of 2H-indazole was accomplished by a Cu(II) salt promoted oxidative cyclization, while 2-substituted indoles could be accessed via a Ag(I) salt mediated N–N bond cleavage. This method represents the first synthesis of indoles from alkynyl triazenes. Computational analysis was performed for both reaction pathways, supporting a Lewis acid role for Cu and a π-acid catalysis for Ag.
Co-reporter:Jiean Chen, Sixuan Meng, Leming Wang, Hongmei Tang and Yong Huang
Chemical Science (2010-Present) 2015 - vol. 6(Issue 7) pp:NaN4189-4189
Publication Date(Web):2015/04/23
DOI:10.1039/C5SC00878F
We report the first asymmetric sulfa-Michael addition (SMA) reactions using a chiral N-heterocyclic carbene (NHC) as a non-covalent organocatalyst. We demonstrate that a triazolium salt derived NHC functions as an excellent Brønsted base to promote enantioselective carbon–sulfur bond formation. The reaction is applicable to a wide range of thiols and electrophilic olefins. Notably, quaternary chiral centers bearing both an S atom and a CF3 group were synthesized with excellent asymmetric control. Mechanistic studies suggest that the facial discrimination is likely to be guided by non-covalent interactions: hydrogen bonding and π–π stacking.
Co-reporter:Tao Cheng, Weiyu Yin, Yi Zhang, Yingnan Zhang and Yong Huang
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 9) pp:NaN1411-1411
Publication Date(Web):2013/12/20
DOI:10.1039/C3OB42196A
A general palladium catalyzed acetoxylation of benzylic C–H bonds has been developed. Picolinamides serve as an excellent directing group for the C–H activation of benzylic methyls. A wide range of 2-amino benzyl alcohol analogues were synthesized in good yields. The products demonstrated broad synthetic utilities toward various benzo-fused heterocycles. Mechanistic studies revealed the key rate-limiting C–H insertion step, which could be affected by the substitution pattern of the parent arene.
Co-reporter:Qian Shao, Jiean Chen, Meihua Tu, David W. Piotrowski and Yong Huang
Chemical Communications 2013 - vol. 49(Issue 94) pp:NaN11100-11100
Publication Date(Web):2013/10/09
DOI:10.1039/C3CC46757K
An enantioselective organocatalytic process for the one-step synthesis of poly-substituted 1,2,4-triazolines is reported. The heterocycle formation is believed to go through a step-wise mechanism of nucleophilic addition of an azlactone to an azodicarboxylate in the presence of an organic base catalyst, followed by a TMSCHN2 mediated heterocyclization. Both theoretical calculations and experimental evidence suggest the pre-organization of the transition state for the chirality determining step via a unique 7-membered intramolecular hydrogen bonding.
Co-reporter:Huan Sun, Nicolas Guimond and Yong Huang
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 36) pp:NaN8397-8397
Publication Date(Web):2016/08/03
DOI:10.1039/C6OB01258B
Transition metal-catalyzed C–H bond insertion is one of the most straightforward strategies to introduce functionalities within a hydrocarbon microenvironment. For the past two decades, selective activation and functionalization of certain inert C–H bonds have been made possible with the help of directing groups (DGs). Despite the enormous advances in the field, an overwhelming majority of systems require two extra steps from their simple precursors: installation and removal of the DGs. Recently, traceless and multitasking groups were invented as a partial solution to DG release. However, installation remains largely unsolved. Ideally, a transient, catalytic DG would circumvent this problem and increase the step- and atom-economy of C–H functionalization processes. In this review, we summarize the recent development of the transient tethering strategy for C–H activation reactions.
Co-reporter:Qi Zhang, Hai-Zhu Yu, Yi-Tong Li, Lei Liu, Yong Huang and Yao Fu
Dalton Transactions 2013 - vol. 42(Issue 12) pp:NaN4184-4184
Publication Date(Web):2013/02/06
DOI:10.1039/C3DT31898B
A systematic theoretical study on the Rh-catalyzed oxidative Heck-coupling of phenol carbamates with alkenes is carried out. Two possible mechanisms (i.e. arene activation-first and alkene activation-first mechanisms) are examined. As to the C–H activation step, four mechanisms including oxidative addition, electrophilic substitution, concerted metallation-deprotonation (CMD), and σ-bond metathesis are evaluated. The calculation results indicate that the arene activation-first mechanism is more favorable for the overall catalytic cycle. This mechanism involves three steps: arene C–H activation at the position ortho to the carbamate directing group affording a six-membered rhodiacycle intermediate, insertion of the alkene double bond into the Rh(III)–aryl bond, and a final β-H elimination step to release the product and re-generate the catalyst. The rate determining step of the overall catalytic cycle is the arene C–H activation step, which is found to proceed through the acetate-assisted CMD mechanism.

![N-[3,5-Bis(trifluoromethyl)phenyl]-N-[(8a,9S)-6-methoxy-9-cinchonanyl]thiourea](/data/chemimg/103500/852913-16-7.png)
![N-[3,5-Bis(trifluoromethyl)phenyl]-N-[(8a,9S)-6-methoxy-9-cinchonanyl]thiourea](/data/chemimg/103500/852913-16-7_b.png)







![2-Penten-1-ol, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/158300/158213-53-7.png)
![2-Penten-1-ol, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/158300/158213-53-7_b.png)








![BENZAMIDE, N-[2-(DIPHENYLPHOSPHINO)PHENYL]-](http://img.cochemist.com/ccimg/91500/91409-99-3.png)
![BENZAMIDE, N-[2-(DIPHENYLPHOSPHINO)PHENYL]-](http://img.cochemist.com/ccimg/91500/91409-99-3_b.png)






![N-[2-(methylsulfanyl)phenyl]benzamide](http://img.cochemist.com/ccimg/52000/51942-32-6.png)
![N-[2-(methylsulfanyl)phenyl]benzamide](http://img.cochemist.com/ccimg/52000/51942-32-6_b.png)
































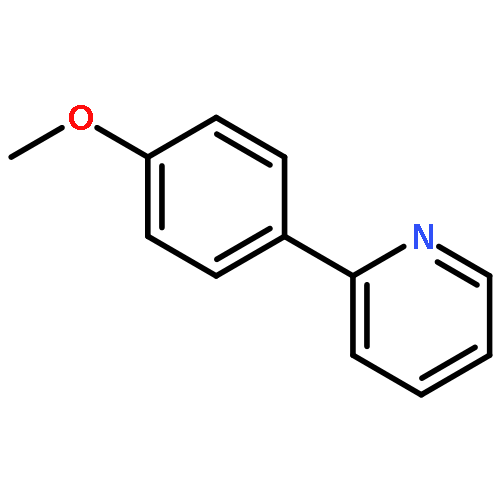
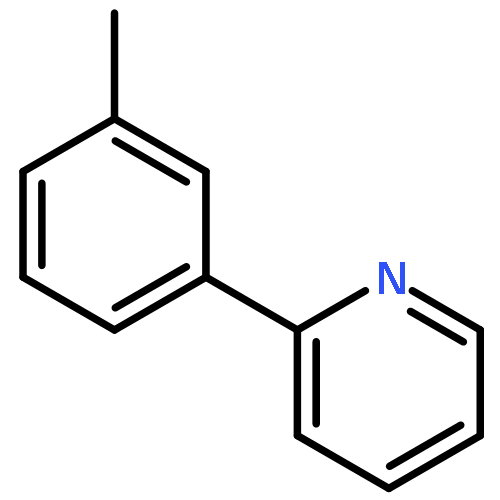
![Acetamide, N-[4-(2-pyridinyl)phenyl]-](/data/chemimg/3753400/860542-25-2.png)
![Acetamide, N-[4-(2-pyridinyl)phenyl]-](/data/chemimg/3753400/860542-25-2_b.png)




![Glycine, N-[1-(2,4-dimethoxyphenyl)-2-[(triphenylmethyl)thio]ethyl]-](http://img.cochemist.com/ccimg/823900/823829-31-8.png)
![Glycine, N-[1-(2,4-dimethoxyphenyl)-2-[(triphenylmethyl)thio]ethyl]-](http://img.cochemist.com/ccimg/823900/823829-31-8_b.png)
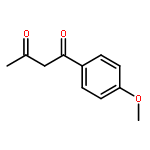
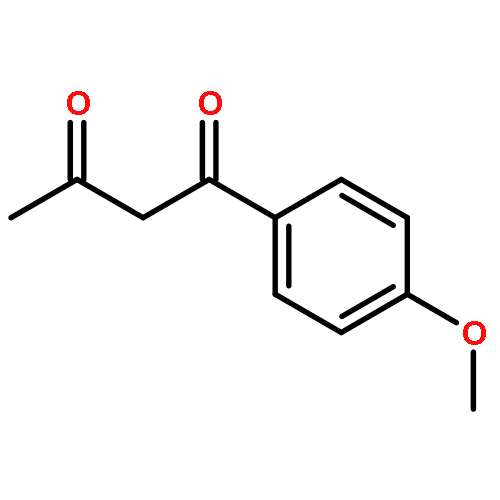
![Ethanone, 1-(2,4-dimethoxyphenyl)-2-[(triphenylmethyl)thio]-](http://img.cochemist.com/ccimg/823900/823829-27-2.png)
![Ethanone, 1-(2,4-dimethoxyphenyl)-2-[(triphenylmethyl)thio]-](http://img.cochemist.com/ccimg/823900/823829-27-2_b.png)















![1-NONYN-3-ONE, 1-[TRIS(1-METHYLETHYL)SILYL]-](http://img.cochemist.com/ccimg/357500/357429-54-0.png)
![1-NONYN-3-ONE, 1-[TRIS(1-METHYLETHYL)SILYL]-](http://img.cochemist.com/ccimg/357500/357429-54-0_b.png)











![2-Pyrrolidinone, 1-[(2E)-1-oxo-2-butenyl]-](http://img.cochemist.com/ccimg/190800/190734-66-8.png)
![2-Pyrrolidinone, 1-[(2E)-1-oxo-2-butenyl]-](http://img.cochemist.com/ccimg/190800/190734-66-8_b.png)
![2,4-Pentanedione, 3-[2-nitro-1-(2-thienyl)ethyl]-, (+)-](http://img.cochemist.com/ccimg/190000/189952-28-1.png)
![2,4-Pentanedione, 3-[2-nitro-1-(2-thienyl)ethyl]-, (+)-](http://img.cochemist.com/ccimg/190000/189952-28-1_b.png)




![1-Pentyn-3-one, 4-methyl-1-[tris(1-methylethyl)silyl]-](http://img.cochemist.com/ccimg/183900/183852-51-9.png)
![1-Pentyn-3-one, 4-methyl-1-[tris(1-methylethyl)silyl]-](http://img.cochemist.com/ccimg/183900/183852-51-9_b.png)
![1-[2-tri(propan-2-yl)silylethynyl]-1λ3,2-benziodoxol-3-one](/data/chemimg/1772700/181934-30-5.png)
![1-[2-tri(propan-2-yl)silylethynyl]-1λ3,2-benziodoxol-3-one](/data/chemimg/1772700/181934-30-5_b.png)

![6H-Pyrrolo[3,2,1-ij]quinolin-6-one, 4,5-dihydro-1,2-diphenyl-](http://img.cochemist.com/ccimg/181400/181305-82-8.png)
![6H-Pyrrolo[3,2,1-ij]quinolin-6-one, 4,5-dihydro-1,2-diphenyl-](http://img.cochemist.com/ccimg/181400/181305-82-8_b.png)


![Benzene, [(3E)-4-nitro-3-butenyl]-](http://img.cochemist.com/ccimg/169800/169775-70-6.png)
![Benzene, [(3E)-4-nitro-3-butenyl]-](http://img.cochemist.com/ccimg/169800/169775-70-6_b.png)














![Benzenesulfonamide, N-[1-(hydroxymethyl)-2-methylpropyl]-4-methyl-,(S)-](http://img.cochemist.com/ccimg/139200/139192-48-6.png)
![Benzenesulfonamide, N-[1-(hydroxymethyl)-2-methylpropyl]-4-methyl-,(S)-](http://img.cochemist.com/ccimg/139200/139192-48-6_b.png)













![Hexanal, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/118800/118794-69-7.png)
![Hexanal, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/118800/118794-69-7_b.png)




![3-[(e)-2-butenoyl]-1,3-oxazolidin-2-one](http://img.cochemist.com/ccimg/109300/109299-92-5.png)
![3-[(e)-2-butenoyl]-1,3-oxazolidin-2-one](http://img.cochemist.com/ccimg/109300/109299-92-5_b.png)


![Benzene, 1-methoxy-4-[(1E)-3-methyl-1-butenyl]-](http://img.cochemist.com/ccimg/103800/103786-20-5.png)
![Benzene, 1-methoxy-4-[(1E)-3-methyl-1-butenyl]-](http://img.cochemist.com/ccimg/103800/103786-20-5_b.png)


























































![Naphthalene, 1-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/37700/37630-26-5.png)
![Naphthalene, 1-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/37700/37630-26-5_b.png)














![Benzene, [(1Z)-2-fluoroethenyl]-](http://img.cochemist.com/ccimg/20500/20405-78-1.png)
![Benzene, [(1Z)-2-fluoroethenyl]-](http://img.cochemist.com/ccimg/20500/20405-78-1_b.png)
![Benzene, [(1E)-2-fluoroethenyl]-](http://img.cochemist.com/ccimg/20500/20405-77-0.png)
![Benzene, [(1E)-2-fluoroethenyl]-](http://img.cochemist.com/ccimg/20500/20405-77-0_b.png)



![Benzene, [(1E)-1-methyl-2-nitroethenyl]-](/data/chemimg/1737100/15241-24-4.png)
![Benzene, [(1E)-1-methyl-2-nitroethenyl]-](/data/chemimg/1737100/15241-24-4_b.png)






![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)

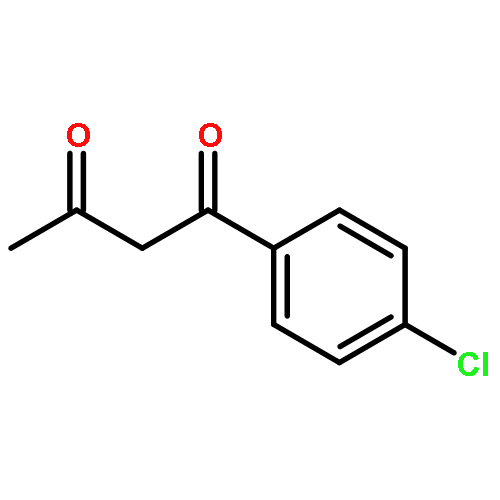
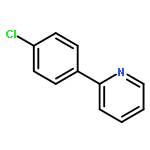
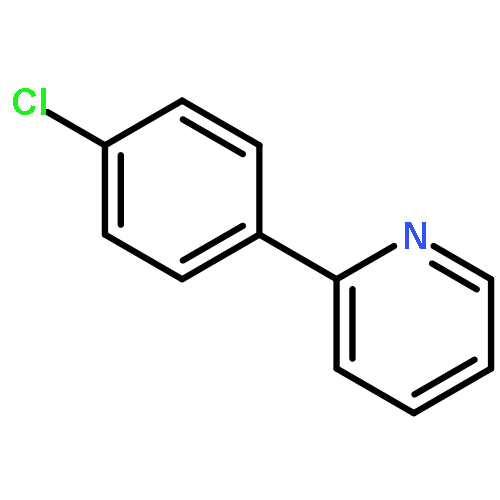
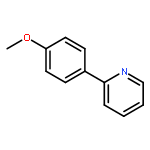


















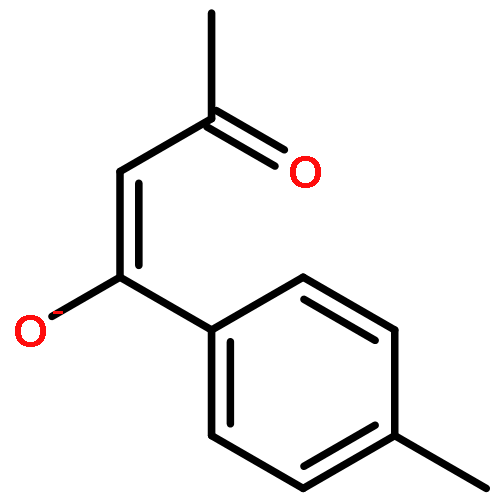


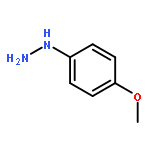
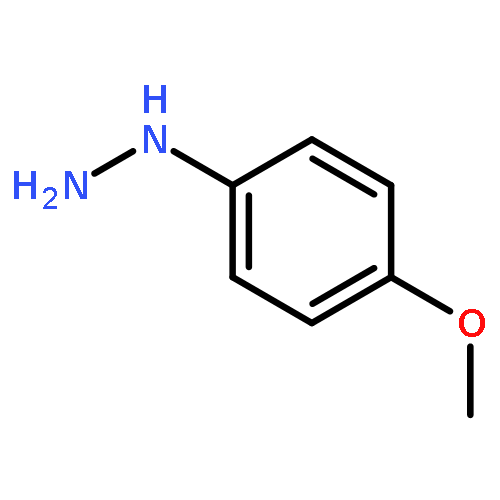
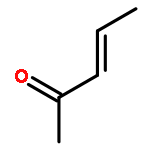
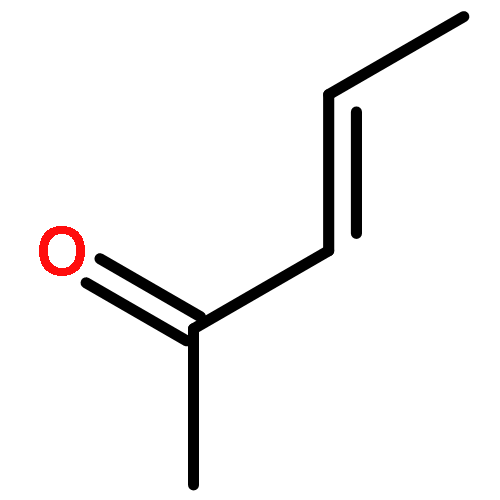
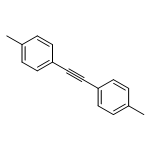
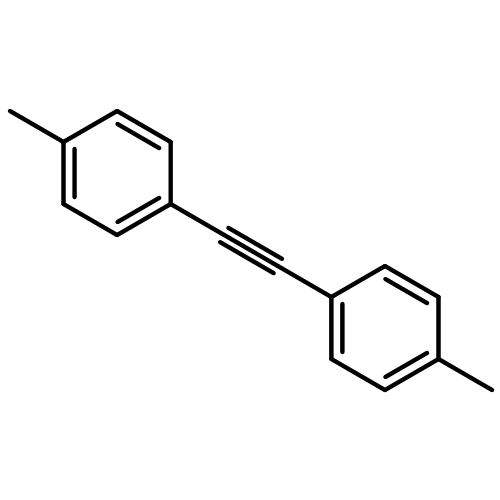






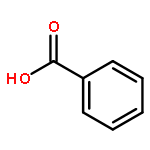
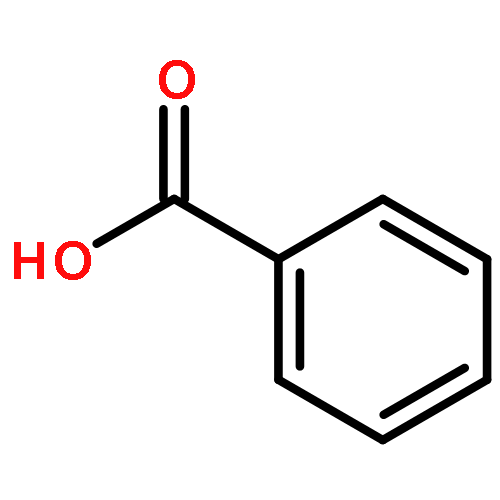
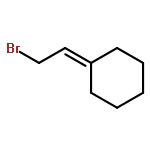

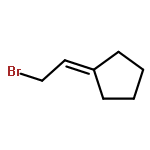
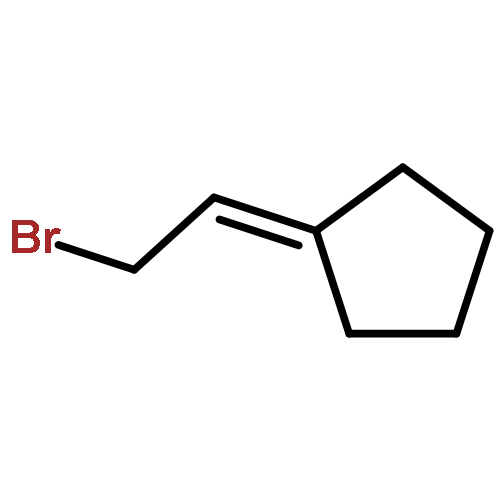
![1,1'-[(Z)-1-fluoroethene-1,2-diyl]dibenzene](http://img.cochemist.com/ccimg/700/671-18-1.png)
![1,1'-[(Z)-1-fluoroethene-1,2-diyl]dibenzene](http://img.cochemist.com/ccimg/700/671-18-1_b.png)





![Benzene, 1-methoxy-2-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/21300/21298-69-1.png)
![Benzene, 1-methoxy-2-[(1E)-2-nitroethenyl]-](http://img.cochemist.com/ccimg/21300/21298-69-1_b.png)