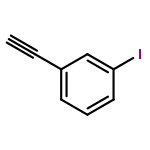
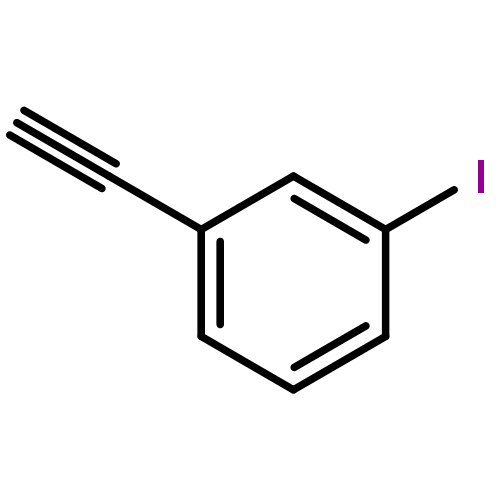
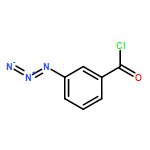
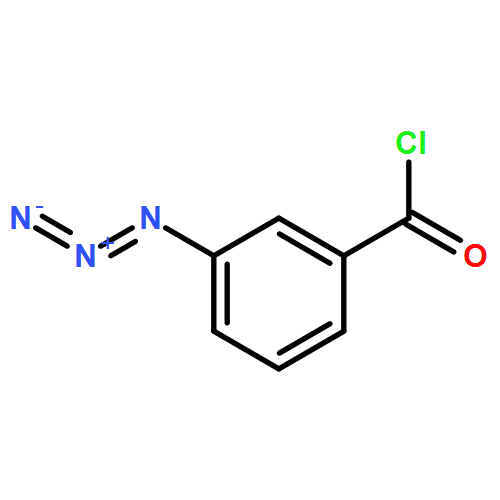
Co-reporter:Yan Li, Zhihai Liu, and Renxiao Wang
Journal of Chemical Information and Modeling September 27, 2010 Volume 50(Issue 9) pp:
Publication Date(Web):August 9, 2010
DOI:10.1021/ci100036a
The molecular mechanics Poisson−Boltzmann surface area (MM-PB/SA) method has been popular for computing protein−ligand binding free energies in recent years. All previous evaluations of the MM-PB/SA method are based upon computer-generated conformational ensembles, which may be affected by the defective computational methods used for preparing these conformational ensembles. In an attempt to reach more convincing conclusions, we have evaluated the MM-PB/SA method on a set of 24 diverse protein−ligand complexes, each of which has a set of conformations derived from NMR spectroscopy. Our results indicate that both MM-PB/SA and molecular mechanics generalized Born surface area (MM-GB/SA) are able to produce a modest correlation between their results and the experimentally measured binding free energies on our test set. In particular, both MM-PB/SA and MM-GB/SA produced better results by using a representative structure (R = 0.72−0.79) rather than averaging over the conformational ensemble of each given complex (R = 0.61−0.74). A head-to-head comparison with four selected scoring functions (X-Score, PLP, ChemScore, and DrugScore) on the same test set reveals that MM-PB/SA and MM-GB/SA results are marginally better than those produced by scoring funcitons, supporting the value of the MM-PB/SA method. Nevertheless, scoring functions are still more cost-effective options, especially for high-throughput tasks.
Co-reporter:Yan Li; Zhixiong Zhao; Zhihai Liu; Minyi Su;Renxiao Wang
Journal of Chemical Information and Modeling 2016 Volume 56(Issue 2) pp:435-453
Publication Date(Web):January 22, 2016
DOI:10.1021/acs.jcim.5b00691
In structure-based drug design, automated de novo design methods are helpful tools for lead discovery as well as lead optimization. In a previous study ( J. Chem. Inf. Model. 2011, 51, 1474−1491) we reported a new de novo design method, namely, Automatic Tailoring and Transplanting (AutoT&T). It overcomes some intrinsic problems in conventional fragment-based buildup methods. In this study, we describe an upgraded version, namely, AutoT&T2. Structural operations conducted by AutoT&T2 have been largely optimized by introducing several new algorithms. As a result, its overall speed in multiround optimization jobs has been improved by a few thousand fold. With this improvement, it is now practical to conduct structural crossover among multiple lead molecules using AutoT&T2. Three different test cases are described in this study that demonstrate the new features and versatile applications of AutoT&T2. The AutoT&T2 software suite is available to the public. Besides, a Web portal for running AutoT&T2 online is provided at http://www.sioc-ccbg.ac.cn/software/att2 for testing.
Co-reporter:Mi Zhou;Qing Li; Renxiao Wang
ChemMedChem 2016 Volume 11( Issue 8) pp:738-756
Publication Date(Web):
DOI:10.1002/cmdc.201500495
Abstract
Protein molecules often interact with other partner protein molecules in order to execute their vital functions in living organisms. Characterization of protein–protein interactions thus plays a central role in understanding the molecular mechanism of relevant protein molecules, elucidating the cellular processes and pathways relevant to health or disease for drug discovery, and charting large-scale interaction networks in systems biology research. A whole spectrum of methods, based on biophysical, biochemical, or genetic principles, have been developed to detect the time, space, and functional relevance of protein–protein interactions at various degrees of affinity and specificity. This article presents an overview of these experimental methods, outlining the principles, strengths and limitations, and recent developments of each type of method.
Co-reporter:Jie Liu and Renxiao Wang
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 3) pp:475-482
Publication Date(Web):February 3, 2015
DOI:10.1021/ci500731a
Scoring functions are a class of computational methods widely applied in structure-based drug design for evaluating protein–ligand interactions. Dozens of scoring functions have been published since the early 1990s. In literature, scoring functions are typically classified as force-field-based, empirical, and knowledge-based. This classification scheme has been quoted for more than a decade and is still repeatedly quoted by some recent publications. Unfortunately, it does not reflect the recent progress in this field. Besides, the naming convention used for describing different types of scoring functions has been somewhat jumbled in literature, which could be confusing for newcomers to this field. Here, we express our viewpoint on an up-to-date classification scheme and appropriate naming convention for current scoring functions. We propose that they can be classified into physics-based methods, empirical scoring functions, knowledge-based potentials, and descriptor-based scoring functions. We also outline the major difference and connections between different categories of scoring functions.
Co-reporter:Ya-li Li, Xiang-yu Qi, Hui Jiang, Xiao-dong Deng, Yan-ping Dong, Ting-bo Ding, Lu Zhou, Peng Men, Yong Chu, Ren-xiao Wang, Xian-cheng Jiang, De-yong Ye
Bioorganic & Medicinal Chemistry 2015 23(18) pp: 6173-6184
Publication Date(Web):
DOI:10.1016/j.bmc.2015.07.060
Co-reporter:Yan Li, Zhihai Liu, Jie Li, Li Han, Jie Liu, Zhixiong Zhao, and Renxiao Wang
Journal of Chemical Information and Modeling 2014 Volume 54(Issue 6) pp:1700-1716
Publication Date(Web):April 9, 2014
DOI:10.1021/ci500080q
Scoring functions are often applied in combination with molecular docking methods to predict ligand binding poses and ligand binding affinities or to identify active compounds through virtual screening. An objective benchmark for assessing the performance of current scoring functions is expected to provide practical guidance for the users to make smart choices among available methods. It can also elucidate the common weakness in current methods for future improvements. The primary goal of our comparative assessment of scoring functions (CASF) project is to provide a high-standard, publicly accessible benchmark of this type. Our latest study, i.e., CASF-2013, evaluated 20 popular scoring functions on an updated set of protein–ligand complexes. This data set was selected out of 8302 protein–ligand complexes recorded in the PDBbind database (version 2013) through a fairly complicated process. Sample selection was made by considering the quality of complex structures as well as binding data. Finally, qualified complexes were clustered by 90% similarity in protein sequences. Three representative complexes were chosen from each cluster to control sample redundancy. The final outcome, namely, the PDBbind core set (version 2013), consists of 195 protein–ligand complexes in 65 clusters with binding constants spanning nearly 10 orders of magnitude. In this data set, 82% of the ligand molecules are “druglike” and 78% of the protein molecules are validated or potential drug targets. Correlation between binding constants and several key properties of ligands are discussed. Methods and results of the scoring function evaluation will be described in a companion work in this issue (doi: 10.1021/ci500081m).
Co-reporter:Yan Li, Li Han, Zhihai Liu, and Renxiao Wang
Journal of Chemical Information and Modeling 2014 Volume 54(Issue 6) pp:1717-1736
Publication Date(Web):April 7, 2014
DOI:10.1021/ci500081m
Our comparative assessment of scoring functions (CASF) benchmark is created to provide an objective evaluation of current scoring functions. The key idea of CASF is to compare the general performance of scoring functions on a diverse set of protein–ligand complexes. In order to avoid testing scoring functions in the context of molecular docking, the scoring process is separated from the docking (or sampling) process by using ensembles of ligand binding poses that are generated in prior. Here, we describe the technical methods and evaluation results of the latest CASF-2013 study. The PDBbind core set (version 2013) was employed as the primary test set in this study, which consists of 195 protein–ligand complexes with high-quality three-dimensional structures and reliable binding constants. A panel of 20 scoring functions, most of which are implemented in main-stream commercial software, were evaluated in terms of “scoring power” (binding affinity prediction), “ranking power” (relative ranking prediction), “docking power” (binding pose prediction), and “screening power” (discrimination of true binders from random molecules). Our results reveal that the performance of these scoring functions is generally more promising in the docking/screening power tests than in the scoring/ranking power tests. Top-ranked scoring functions in the scoring power test, such as X-ScoreHM, ChemScore@SYBYL, ChemPLP@GOLD, and PLP@DS, are also top-ranked in the ranking power test. Top-ranked scoring functions in the docking power test, such as ChemPLP@GOLD, Chemscore@GOLD, GlidScore-SP, LigScore@DS, and PLP@DS, are also top-ranked in the screening power test. Our results obtained on the entire test set and its subsets suggest that the real challenge in protein–ligand binding affinity prediction lies in polar interactions and associated desolvation effect. Nonadditive features observed among high-affinity protein–ligand complexes also need attention.
Co-reporter:Xiaodong Deng, Fu Lin, Ya Zhang, Yan Li, Lu Zhou, Bin Lou, Yue Li, Jibin Dong, Tingbo Ding, Xiancheng Jiang, Renxiao Wang, Deyong Ye
European Journal of Medicinal Chemistry 2014 Volume 73() pp:1-7
Publication Date(Web):12 February 2014
DOI:10.1016/j.ejmech.2013.12.002
•Sphingomyelin synthase is a novel potential drug target for metabolic diseases.•The small-molecule SMS inhibitors discovered by rational design are firstly revealed.•The hit D2 was demonstrated to be an effective SMS inhibitor both in vitro and in vivo.Sphingomyelin synthase (SMS), which catalyzes ceramide as one of the substrates to produce sphingomyelin, is a critical factor in the sphingolipid biosynthesis pathway. Recent studies indicated that SMS could serve as a novel potential drug target for the treatment of various metabolic diseases such as insulin resistance and atherosclerosis. However, very few small-molecule inhibitors of SMS are known. In this study, we performed structure-based virtual screening in combination with chemical synthesis and bioassay and discovered a class of small-molecule SMS inhibitors. The most potent compound exhibited an IC50 value lower than 20 μM in an in vitro enzymatic assay. To the best of our knowledge, this is the first time that small-molecule SMS inhibitors with potency close to the micromolar range are publicly revealed. The structure–activity relationship demonstrated by this class of compounds provides insights into the structural features that are essential for effective SMS inhibition.The hit D2 was supposed be a potential molecular tool in SMS bio-function studies as well as for developing a new class of therapeutic drug treating various metabolic diseases.
Co-reporter:Dr. Chengwen Yang;Sha Chen;Mi Zhou;Dr. Yan Li;Yangfeng Li;Zhengxi Zhang;Dr. Zhen Liu;Dr. Qian Ba;Dr. Jingquan Li; Hui Wang; Xiaomei Yan; Dawei Ma; Renxiao Wang
ChemMedChem 2014 Volume 9( Issue 7) pp:1436-1452
Publication Date(Web):
DOI:10.1002/cmdc.201400058
Abstract
Antiapoptotic Bcl-2 family proteins, such as Bcl-xL, Bcl-2, and Mcl-1, are often overexpressed in tumor cells, which contributes to tumor cell resistance to chemotherapies and radiotherapies. Inhibitors of these proteins thus have potential applications in cancer treatment. We discovered, through structure-based virtual screening, a lead compound with micromolar binding affinity to Mcl-1 (inhibition constant (Ki)=3 μM). It contains a phenyltetrazole and a hydrazinecarbothioamide moiety, and it represents a structural scaffold not observed among known Bcl-2 inhibitors. This work presents the structural optimization of this lead compound. By following the scaffold-hopping strategy, we have designed and synthesized a total of 82 compounds in three sets. All of the compounds were evaluated in a fluorescence-polarization binding assay to measure their binding affinities to Bcl-xL, Bcl-2, and Mcl-1. Some of the compounds with a 3-phenylthiophene-2-sulfonamide core moiety showed sub-micromolar binding affinities to Mcl-1 (Ki=0.3–0.4 μM) or Bcl-2 (Ki≈1 μM). They also showed obvious cytotoxicity on tumor cells (IC50<10 μM). Two-dimensional heteronuclear single quantum coherence NMR spectra of three selected compounds, that is, YCW-E5, YCW-E10, and YCW-E11, indicated that they bind to the BH3-binding groove on Bcl-xL in a similar mode to ABT-737. Several apoptotic assays conducted on HL-60 cells demonstrated that these compounds are able to induce cell apoptosis through the mitochondrial pathway. We propose that the compounds with the 3-phenylthiophene-2-sulfonamide core moiety are worth further optimization as effective apoptosis inducers with an interesting selectivity towards Mcl-1 and Bcl-2.
Co-reporter:Yan Li, Zhihai Liu, Li Han, Chengke Li, and Renxiao Wang
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 9) pp:2437-2447
Publication Date(Web):August 10, 2013
DOI:10.1021/ci400241s
Protein–protein interactions are observed in various biological processes. They are important for understanding the underlying molecular mechanisms and can be potential targets for developing small-molecule regulators of such processes. Previous studies suggest that certain residues on protein–protein binding interfaces are ″hot spots″. As an extension to this concept, we have developed a residue-based method to identify the characteristic interaction patterns (CIPs) on protein–protein binding interfaces, in which each pattern is a cluster of four contacting residues. Systematic analysis was conducted on a nonredundant set of 1,222 protein–protein binding interfaces selected out of the entire Protein Data Bank. Favored interaction patterns across different protein–protein binding interfaces were retrieved by considering both geometrical and chemical conservations. As demonstrated on two test tests, our method was able to predict hot spot residues on protein–protein binding interfaces with good recall scores and acceptable precision scores. By analyzing the function annotations and the evolutionary tree of the protein–protein complexes in our data set, we also observed that protein–protein interfaces sharing common characteristic interaction patterns are normally associated with identical or similar biological functions.
Co-reporter:Yaochun Xu;Mi Zhou;Yan Li;Chengke Li;Zhengxi Zhang; Biao Yu; Renxiao Wang
ChemMedChem 2013 Volume 8( Issue 8) pp:1345-1352
Publication Date(Web):
DOI:10.1002/cmdc.201300159
Abstract
In a previous study we reported a class of compounds with a 2H-thiazolo[3,2-a]pyrimidine core structure as general inhibitors of anti-apoptotic Bcl-2 family proteins. However, the absolute stereochemical configuration of one carbon atom on the core structure remained unsolved, and its potential impact on the binding affinities of compounds in this class was unknown. In this study, we obtained pure R and S enantiomers of four selected compounds by HPLC separation and chiral synthesis. The absolute configurations of these enantiomers were determined by comparing their circular dichroism spectra to that of an appropriate reference compound. In addition, a crystal structure of one selected compound revealed the exocyclic double bond in these compounds to be in the Z configuration. The binding affinities of all four pairs of enantiomers to Bcl-xL, Bcl-2, and Mcl-1 proteins were measured in a fluorescence-polarization-based binding assay, yielding inhibition constants (Ki values) ranging from 0.24 to 2.20 μM. Interestingly, our results indicate that most R and S enantiomers exhibit similar binding affinities for the three tested proteins. A binding mode for this compound class was derived by molecular docking and molecular dynamics simulations to provide a reasonable interpretation of this observation.
Co-reporter:Mi Zhou ; Renxiao Wang
ChemMedChem 2013 Volume 8( Issue 5) pp:694-707
Publication Date(Web):
DOI:10.1002/cmdc.201200560
Abstract
Autophagy is a highly conserved process in which damaged proteins and organelles are sequestered in double-membrane autophagosomes and delivered to lysosomes for degradation and recycling. As an efficient response to cellular stress, autophagy is essential for the maintenance of cellular homeostasis. Defective autophagy is associated with a variety of diseases, including cancer. This article summarizes current knowledge about the molecular mechanism of autophagy and its role in tumorigenesis. Particular focus is placed on the development of small-molecule regulators of autophagy and their potential application as anticancer therapeutic agents.
Co-reporter:Dr. Xiao Ding;Dr. Yan Li;Li Lv;Mi Zhou;Dr. Li Han;Zhengxi Zhang;Dr. Qian Ba;Dr. Jingquan Li; Hui Wang; Hong Liu; Renxiao Wang
ChemMedChem 2013 Volume 8( Issue 12) pp:1986-2014
Publication Date(Web):
DOI:10.1002/cmdc.201300316
Abstract
Considerable efforts have been made to the development of small-molecule inhibitors of antiapoptotic B-cell lymphoma 2 (Bcl-2) family proteins (such as Bcl-2, Bcl-xL, and Mcl-1) as a new class of anticancer therapies. Unlike general inhibitors of the entire family, selective inhibitors of each member protein can hopefully reduce the adverse side effects in chemotherapy treatments of cancers overexpressing different Bcl-2 family proteins. In this study, we designed four series of benzylpiperazine derivatives as plausible Bcl-2 inhibitors based on the outcomes of a computational algorithm. A total of 81 compounds were synthesized, and their binding affinities to Bcl-2, Bcl-xL, and Mcl-1 measured. Encouragingly, 22 compounds exhibited binding affinities in the micromolar range (Ki<20 μM) to at least one target protein. Moreover, some compounds were observed to be highly selective binders to Mcl-1 with no detectable binding to Bcl-2 or Bcl-xL, among which the most potent one has a Ki value of 0.18 μM for Mcl-1. Binding modes of four selected compounds to Mcl-1 and Bcl-xL were derived through molecular docking and molecular dynamics simulations. It seems that the binding affinity and selectivity of these compounds can be reasonably interpreted with these models. Our study demonstrated the possibility for obtaining selective Mcl-1 inhibitors with relatively simple chemical scaffolds. The active compounds identified by us could be used as lead compounds for developing even more potent selective Mcl-1 inhibitors with potential pharmaceutical applications.
Co-reporter:Yan Li, Yuan Zhao, Zhihai Liu, and Renxiao Wang
Journal of Chemical Information and Modeling 2011 Volume 51(Issue 6) pp:1474-1491
Publication Date(Web):April 26, 2011
DOI:10.1021/ci200036m
Docking-based virtual screening of large compound libraries has been widely applied to lead discovery in structure-based drug design. However, subsequent lead optimizations often rely on other types of computational methods, such as de novo design methods. We have developed an automatic method, namely automatic tailoring and transplanting (AutoT&T), which can effectively utilize the outcomes of virtual screening in lead optimization. This method detects suitable fragments on virtual screening hits and then transplants them onto a lead compound to generate new ligand molecules. Binding affinities, synthetic feasibilities, and drug-likeness properties are considered in the selection of final designs. In this study, our AutoT&T program was tested on three different target proteins, including p38 MAP kinase, PPAR-α, and Mcl-1. In the first two cases, AutoT&T was able to produce molecules identical or similar to known inhibitors with better potency than the given lead compound. In the third case, we demonstrated how to apply AutoT&T to design novel ligand molecules from scratch. Compared to the solutions generated by other two de novo design methods, i.e., LUDI and EA-Inventor, the solutions generated by AutoT&T were structurally more diverse and more promising in terms of binding scores in all three cases. AutoT&T also completed the assigned jobs more efficiently than LUDI and EA-Inventor by several folds. Our AutoT&T method has certain technical advantages over de novo design methods. Importantly, it expands the application of virtual screening from lead discovery to lead optimization and thus may serve as a valuable tool for many researchers.
Co-reporter:Jiangping Lou, Zhen Liu, Yan Li, Mi Zhou, Zhengxi Zhang, Shu Zheng, Renxiao Wang, Jian Li
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 22) pp:6662-6666
Publication Date(Web):15 November 2011
DOI:10.1016/j.bmcl.2011.09.061
A compound with a cyclic thienopyrimidine moiety and an aceto-hydrazone moiety in its chemical structure was discovered in a cell-based screening to have noticeable cytotoxicity on several tumor cell lines. A total of 38 derivatives of this compound were synthesized at five steps with high yields. These compounds were tested in standard MTT assays, and several compounds exhibited improved cytotoxic activities. The most potent compounds have IC50 values of 10–20 μM on A549, HeLa, and MBA-MD-231 tumor cells. Flow cytometry analysis of several active compounds and subsequent examination of caspase activation indicate that they induce caspase-dependent apoptosis in tumor cells. In addition, these compounds do not have obvious effect on a normal cell line HEK-293T, demonstrating the desired selectivity against tumor cells. Results from a fluorescence polarization-based in vitro binding assay indicate that this class of compounds does not significantly interrupt the interactions between Mcl-1 and Bid. Their cytotoxicity is achieved presumably through other mechanisms.IC50 = 10–20 μM on A549, Hela and MDA-MB-231 tumor cells.
Co-reporter:Ya Zhang;Fu Lin;Xiaodong Deng;Renxiao Wang;Deyong Ye
Chinese Journal of Chemistry 2011 Volume 29( Issue 8) pp:1567-1575
Publication Date(Web):
DOI:10.1002/cjoc.201180282
Abstract
Sphingomyelin synthase (SMS) produces sphingomyelin and diacylglycerol from ceramide and phosphatidylcholine. It plays an important role in cell survival and apoptosis, inflammation, and lipid homeostasis, and therefore has been noticed in recent years as a novel potential drug target. In this study, we combined homology modeling, molecular docking, molecular dynamics simulation, and normal mode analysis to derive a three-dimensional structure of human sphingomyelin synthase (hSMS1) in complex with sphingomyelin. Our model provides a reasonable explanation on the catalytic mechanism of hSMS1. It can also explain the high selectivity of hSMS1 towards phosphocholine and sphingomyelin as well as some other known experimental results about hSMS1. Moreover, we also derived a complex model of D609, the only known small-molecule inhibitor of hSMS1 so far. Our hSMS1 model may serve as a reasonable structural basis for the discovery of more effective small-molecule inhibitors of hSMS1.
Co-reporter:Bingcheng Zhou;Dr. Xun Li;Dr. Yan Li;Yaochun Xu;Zhengxi Zhang;Mi Zhou;Dr. Xinglong Zhang;Dr. Zhen Liu; Jiahai Zhou; Chunyang Cao; Biao Yu; Renxiao Wang
ChemMedChem 2011 Volume 6( Issue 5) pp:904-921
Publication Date(Web):
DOI:10.1002/cmdc.201000484
Abstract
A class of compounds with a common thiazolo[3,2-a]pyrimidinone motif has been developed as general inhibitors of Bcl-2 family proteins. The lead compound was originally identified in a random screening of a small compound library using a fluorescence polarization-based competitive binding assay. Its binding to the Bcl-xL protein was further confirmed by 15N-HSQC NMR experiments. Structural modifications on the lead compound were guided by the outcomes of molecular modeling studies. Among the 42 compounds obtained, a number of them exhibited much improved binding affinities to Bcl-2 family proteins as compared to the lead compound. The most potent compound, BCL-LZH-40, inhibited the binding of BH3 peptides to Bcl-xL, Bcl-2, and Mcl-1 with inhibition constants (Ki) of 17, 534, and 200 nM, respectively.
Co-reporter:Fu Lin and Renxiao Wang
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 6) pp:1852-1870
Publication Date(Web):May 10, 2010
DOI:10.1021/ct900454q
Metal ions are indispensable for maintaining the structural stability and catalytic activity of metalloproteins. Molecular modeling studies of such proteins with force fields, however, are often hampered by the “missing parameter” problem. In this study, we have derived bond-stretching and angle-bending parameters applicable to zinc-containing systems which are compatible with the AMBER force field. A total of 18 model systems were used to mimic the common coordination configurations observed in the complexes formed by zinc-containing metalloproteins. The Hessian matrix of each model system computed at the B3LYP/6-311++G(2d,2p) level was then analyzed by Seminario’s method to derive the desired force constants. These parameters were validated extensively in structural optimizations and molecular dynamics simulations of four selected model systems as well as one protein−ligand complex formed by carbonic anhydrase II. The best performance was achieved by a bonded model in combination with the atomic partial charges derived by the restrained electrostatic potential method. After some minor optimizations, this model was also able to reproduce the vibrational frequencies computed by quantum mechanics. This study provides a comprehensive set of force field parameters applicable to a variety of zinc-containing molecular systems. In principle, our approach can be applied to other molecular systems with missing force field parameters.
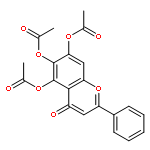
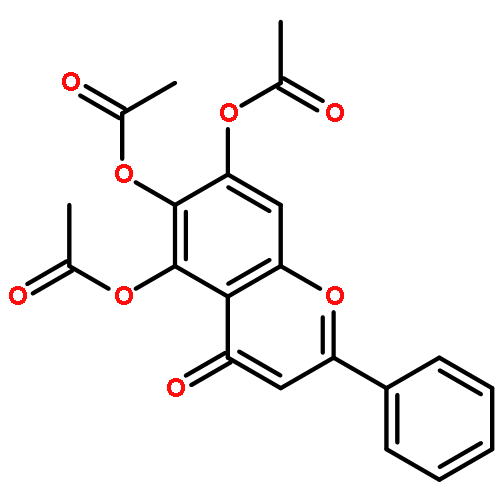
Co-reporter:Hefang Shi, Bingcheng Zhou, Wenwen Li, Zhimin Shi, Biao Yu, Renxiao Wang
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 9) pp:2855-2858
Publication Date(Web):1 May 2010
DOI:10.1016/j.bmcl.2010.03.045
A synthetic method of introducing bulky aryl groups at the 2-O- and 6-O-positions on glucopyranosides was developed. A total of 37 new compounds of this class were obtained successfully. These compounds were tested on several tumor cell lines by MTT assays, and some of them exhibited encouraging inhibitory activities. The most potent compound, CAB-SHZH-27, exhibited EC50 values of 14, 12, and 10 μmol/L on A549, MDA-MB-231 and HeLa cells, respectively. A preliminary structure–activity relationship analysis indicates that the two free hydroxyl groups on the d-glucose core are indispensable for the biological activities of this class of compounds, and the aryl group at the 6-O-position has a more obvious impact than the one at the 2-O-position. An interesting ‘on–off’ mechanism of this class of compounds was also observed in our MTT assays, which remains to be explored.Structure of CAB-SHZH27 and its dose-dependent cytotoxicity on MDA-MB-231 cells.
Co-reporter:Fu Lin;Renxiao Wang
Journal of Molecular Modeling 2010 Volume 16( Issue 1) pp:107-118
Publication Date(Web):2010 January
DOI:10.1007/s00894-009-0523-0
Saponins are a class of compounds containing a triterpenoid or steroid core with some attached carbohydrate modules. Many saponins cause hemolysis. However, the hemolytic mechanism of saponins at the molecular level is not yet fully understood. In an attempt to explore this issue, we have studied dioscin—a saponin with high hemolytic activity—through extensive molecular dynamics (MD) simulations. Firstly, all-atom MD simulations of 8 ns duration were conducted to study the stability of the dioscin–cholesterol complex and the cholesterol–cholesterol complex in water and in decane, respectively. MM-GB/SA computations indicate that the dioscin–cholesterol complex is energetically more favorable than the cholesterol–cholesterol complex in a non-polar environment. Next, several coarse-grained MD simulations of 400 ns duration were conducted to directly observe the distribution of multiple dioscin molecules on a DPPC-POPC-PSM-CHOL lipid bilayer. Our results indicate that dioscin can penetrate into the lipid bilayer, accumulate in the lipid raft micro-domain, and then bind cholesterol. This leads to the destabilization of lipid raft and consequent membrane curvature, which may eventually result in the hemolysis of red cells. This possible mechanism of hemolysis can well explain some experimental observations on hemolysis.
Co-reporter:Xinglong Zhang, Xun Li and Renxiao Wang
Journal of Chemical Information and Modeling 2009 Volume 49(Issue 4) pp:1033-1048
Publication Date(Web):March 25, 2009
DOI:10.1021/ci8004429
We have studied the binding affinities of a set of 45 small-molecule inhibitors to protein tyrosine phosphatase 1B (PTP1B) through computational approaches. All of these compounds share a common oxalylamino benzoic acid (OBA) moiety. The complex structure of each compound was modeled by using the GOLD program plus the ASP scoring function. Each complex structure was then subjected to a molecular dynamics (MD) simulation of 2 ns long by using the AMBER program. Based on the configurational ensembles retrieved from MD trajectories, both MM-GB/SA and MM-PB/SA were employed to compute the binding free energies of all 45 PTP1B inhibitors. The correlation coefficient between the MM-GB/SA results and experimental binding data was 0.87 and the standard deviation was 0.60 kcal/mol. The performance of MM-PB/SA was slightly inferior to that of MM-GB/SA. Several aspects of the MM-GB(PB)/SA method were explored in our study to obtain optimized results. The X-Score scoring function was found to produce equally good results as MM-GB/SA on both the complex structures prepared by molecular docking and the configurational ensembles obtained through lengthy MD simulations. The structure−activity relationship of this set of compounds is also discussed based on the computed results. The computational approaches validated in our study are hopefully applicable to the study of other classes of PTP1B inhibitors.
Co-reporter:Tiejun Cheng, Xun Li, Yan Li, Zhihai Liu and Renxiao Wang
Journal of Chemical Information and Modeling 2009 Volume 49(Issue 4) pp:1079-1093
Publication Date(Web):April 9, 2009
DOI:10.1021/ci9000053
Scoring functions are widely applied to the evaluation of protein−ligand binding in structure-based drug design. We have conducted a comparative assessment of 16 popular scoring functions implemented in main-stream commercial software or released by academic research groups. A set of 195 diverse protein−ligand complexes with high-resolution crystal structures and reliable binding constants were selected through a systematic nonredundant sampling of the PDBbind database and used as the primary test set in our study. All scoring functions were evaluated in three aspects, that is, “docking power”, “ranking power”, and “scoring power”, and all evaluations were independent from the context of molecular docking or virtual screening. As for “docking power”, six scoring functions, including GOLD::ASP, DS::PLP1, DrugScorePDB, GlideScore-SP, DS::LigScore, and GOLD::ChemScore, achieved success rates over 70% when the acceptance cutoff was root-mean-square deviation < 2.0 Å. Combining these scoring functions into consensus scoring schemes improved the success rates to 80% or even higher. As for “ranking power” and “scoring power”, the top four scoring functions on the primary test set were X-Score, DrugScoreCSD, DS::PLP, and SYBYL::ChemScore. They were able to correctly rank the protein−ligand complexes containing the same type of protein with success rates around 50%. Correlation coefficients between the experimental binding constants and the binding scores computed by these scoring functions ranged from 0.545 to 0.644. Besides the primary test set, each scoring function was also tested on four additional test sets, each consisting of a certain number of protein−ligand complexes containing one particular type of protein. Our study serves as an updated benchmark for evaluating the general performance of today’s scoring functions. Our results indicate that no single scoring function consistently outperforms others in all three aspects. Thus, it is important in practice to choose the appropriate scoring functions for different purposes.
Co-reporter:Xun LI ;Renxiao WANG
Chinese Journal of Chemistry 2009 Volume 27( Issue 1) pp:23-28
Publication Date(Web):
DOI:10.1002/cjoc.200990021
Abstract
An automatic method has been developed for identifying antibody entries in the protein data bank (PDB). Our method, called KIAb (Keyword-based Identification of Antibodies), parses PDB-format files to search for particular keywords relevant to antibodies, and makes judgment accordingly. Our method identified 780 entries as antibodies on the entire PDB. Among them, 767 entries were confirmed by manual inspection, indicating a high success rate of 98.3%. Our method recovered basically all of the entries compiled in the Summary of Antibody Crystal Structures (SACS) database. It also identified a number of entries missed by SACS. Our method thus provides a more complete mining of antibody entries in PDB with a very low false positive rate.
Co-reporter:Yan Li, Bingcheng Zhou, Renxiao Wang
Journal of Molecular Graphics and Modelling 2009 Volume 28(Issue 3) pp:203-219
Publication Date(Web):October 2009
DOI:10.1016/j.jmgm.2009.07.001
Neuraminidase is an attractive therapeutic target for standing against influenza virus, such as the threatening avian influenza virus H5N1. A recently discovered cavity near the well-known catalytic site on neuraminidase subtype 1 (N1) provides a good possibility to develop dual-site-binding inhibitors, which may achieve improved activities and selectivities against N1. We have designed some derivatives of Tamiflu with such features through a fragment-based approach combining multiple computational methods. Over 1000 FDA-approved small-molecule drugs were computationally screened targeting at the open conformation of N1 with the GOLD program in combination with the X-Score scoring function. Some chemical fragments on the top-scored hits, which were able to fit into the 150-cavity, were transplanted onto the core structure of Tamiflu to produce a total of 30 new molecules. Then, binding of these designed molecules to N1 was evaluated by molecule docking. The promising ones were further subjected to molecular dynamics simulation of 3 ns long, and their binding free energies were computed by using the MM-PB/SA method. Some of our designed molecules were predicted to have comparable or even better binding affinities than that of Tamiflu. We report our results herein so that other researchers who have the necessary chemical and biological resources can utilize them in the development of new N1 inhibitors. In addition, our study actually suggests a practical strategy for optimizing a given lead compound based on the outcomes of a standard virtual screening trial.
Co-reporter:Fu Lin;Renxiao Wang
Journal of Molecular Modeling 2009 Volume 15( Issue 1) pp:
Publication Date(Web):2009 January
DOI:10.1007/s00894-008-0372-2
Glucagon-like peptide-1 receptor (GLP-1R) is a promising molecular target for developing drugs treating type 2 diabetes. We have predicted the complete three-dimensional structure of GLP-1R and the binding modes of several GLP-1R agonists, including GLP-1, Boc5, and Cpd1, through a combination of homology modeling, molecular docking, and long-time molecular dynamics simulation on a lipid bilayer. Our model can reasonably interpret the results of a number of mutation experiments regarding GLP-1R as well as the successful modification to GLP-1 by Liraglutide. Our model is also validated by a recently revealed crystal structure of the extracellular domain of GLP-1R. An activation mechanism of GLP-1R agonists is proposed based on the principal component analysis and normal mode analysis on our predicted GLP-1R structure. Before the complete structure of GLP-1R is determined through experimental means, our model may serve as a valuable reference for characterizing the interactions between GLP-1R and its agonists.
Co-reporter:Zhiguo Liu, Guitao Wang, Zhanting Li and Renxiao Wang
Journal of Chemical Theory and Computation 2008 Volume 4(Issue 11) pp:1959-1973
Publication Date(Web):October 15, 2008
DOI:10.1021/ct800267x
We have conducted potential of mean force (PMF) analyses to derive the geometrical parameters of various types of hydrogen bonds on protein−ligand binding interface. Our PMF analyses are based on a set of 4535 high-quality protein−ligand complex structures, which are compiled through a systematic mining of the entire Protein Data Bank. Hydrogen bond donor and acceptor atoms are classified into several basic types. Both distance- and angle-dependent statistical potentials are derived for each donor−acceptor pair, from which distance and angle cutoffs are obtained in an objective, unambiguous manner. These donor−acceptor pairs are also studied by quantum mechanics (QM) calculations at the MP2/6−311++G** level on model molecules. Comparison of the outcomes of PMF analyses and QM calculations suggests that QM calculation may serve as an alternative approach for characterizing hydrogen bond geometry. Both of our PMF analyses and QM calculations indicate that C−H···O hydrogen bonds are relatively weak as compared to common hydrogen bonds formed between nitrogen and oxygen atoms. A survey on the protein−ligand complex structures in our data set has revealed that Cα-H···O hydrogen bonds observed in protein−ligand binding are frequently accompanied by bifurcate N−H···O hydrogen bonds. Thus, the Cα-H···O hydrogen bonds in such cases would better be interpreted as secondary interactions.
Co-reporter:Renxiao Wang, Fu Lin, Yong Xu, Tiejun Cheng
Journal of Molecular Graphics and Modelling 2007 Volume 26(Issue 1) pp:368-377
Publication Date(Web):July 2007
DOI:10.1016/j.jmgm.2007.01.006
We have developed a new empirical model, I-SOLV, for computing solvation free energies of neutral organic molecules. It computes the solvation free energy of a solute molecule by summing up the contributions from its component atoms. The contribution from a certain atom is determined by the solvent-accessible surface area as well as the surface tension of this atom. A total of 49 atom types are implemented in our model for classifying C, N, O, S, P, F, Cl, Br and I in common organic molecules. Their surface tensions are parameterized by using a data set of 532 neutral organic molecules with experimentally measured solvation free energies. A head-to-head comparison of our model with several other solvation models was performed on a test set of 82 molecules. Our model outperformed other solvation models, including widely used PB/SA and GB/SA models, with a mean unsigned error as low as 0.39 kcal/mol. Our study has demonstrated again that well-developed empirical solvation models are not necessarily less accurate than more sophisticated theoretical models. Empirical models may serve as appealing alternatives due to their simplicity and accuracy.