Co-reporter:Liping Zhang;Hong Zhang;Dr. Xuehua Zheng;Yining Zhao;Dr. Shangke Chen;Yunyun Chen;Renwei Zhang; Qing Li; Xiaopeng Hu
ChemMedChem 2014 Volume 9( Issue 4) pp:706-709
Publication Date(Web):
DOI:10.1002/cmdc.201300455
Abstract
Caffeic acid phenethyl ester (CAPE), the major bioactive component of honeybee propolis, is a potent selective inhibitor of aldo-keto reductase family member 1B10 (AKR1B10), and a number of derivatives hold promise as potential anticancer agents. However, sequence homology between AKR1B10 and other members of the superfamily, including critical phase I metabolizing enzymes, has resulted in a concern over the selectivity of any potential therapeutic agent. To elucidate the binding mode of CAPE with AKR1B10 and to provide a tool for future in silico efforts towards identifying selective inhibitors, the crystal structure of AKR1B10 in complex with CAPE was determined. The observed interactions provide an explanation for the selectivity exhibited by CAPE for AKR1B10, and could be used to guide further derivative design.
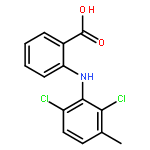
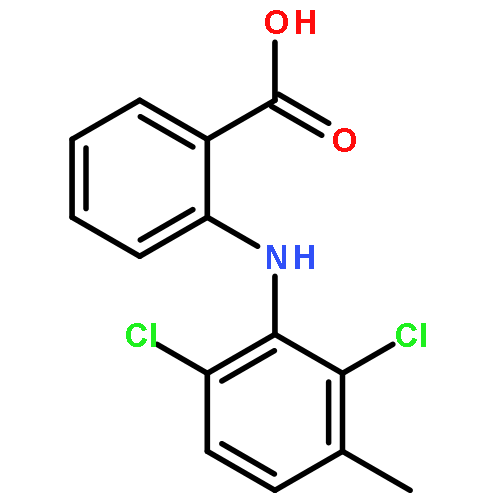
Co-reporter:Jing Zhai;Hong Zhang;Ligping Zhang;Yining Zhao;Dr. Sangke Chen;Yunyun Chen;Xinyu Peng; Qing Li;Dr. Minggui Yuan; Xiaopeng Hu
ChemMedChem 2013 Volume 8( Issue 9) pp:1462-1464
Publication Date(Web):
DOI:10.1002/cmdc.201300243
Co-reporter:Dr. Xuehua Zheng;Liping Zhang;Weijia Chen;Yunyun Chen; Wei Xie; Xiaopeng Hu
ChemMedChem 2012 Volume 7( Issue 11) pp:1921-1923
Publication Date(Web):
DOI:10.1002/cmdc.201200333
Co-reporter:Ming Liu, Minggui Yuan, Zhe Li, Yuen-Kit Cheng, Hai-Bin Luo, Xiaopeng Hu
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 14) pp:4243-4247
Publication Date(Web):15 July 2011
DOI:10.1016/j.bmcl.2011.05.095
In the present work, a combined study of kinetic analysis, molecular docking, and molecular dynamics simulations on indomethacin and its analogues is performed to better understand their inhibitory mechanisms towards human glyoxalase I (GLOI). A remarkable correlation (R2 = 0.974) was observed for six inhibitors including indomethacin between their experimental inhibitory affinities and predicted binding free energy parameter (ΔGbind,pred). This suggests that ΔGbind,pred of a GLOI/inhibitor complex can be efficiently used to interpolate the experimental inhibitory affinity of a ligand of similar nature in the GLOI enzyme system. Energetic analyses revealed that electrostatic contribution plays an important role in their inhibitory mechanisms, which reflects the significant contribution of the coordination bond between zinc and ligands. The present work highlights that indomethacin is a promising lead as GLOI inhibitors for further development since it may bind all subsites in the active site pocket of GLOI and stabilize the flexible loop (152–159).
Co-reporter:Ming Liu, Minggui Yuan, Minxian Luo, Xianzhang Bu, Hai-Bin Luo, Xiaopeng Hu
Biophysical Chemistry 2010 Volume 147(1–2) pp:28-34
Publication Date(Web):March 2010
DOI:10.1016/j.bpc.2009.12.007
Glyoxalase I (GLOI) is a key metalloenzyme in glycolytic pathway by detoxifying reactive α-ketoaldehydes such as methylglyoxal. Recent studies demonstrate that the nature product curcumin is an efficient inhibitor of GLOI, but its binding mechanism towards GLOI is still unclear. In the present study, molecular docking and molecular dynamics (MD) simulations were performed to better understand the inhibitory mechanism of curcumin towards GLOI. The enol form of curcumin coordinates with the catalytic zinc ion of GLOI and forms a strong hydrogen bond with Glu 172, whereas its keto tautomer displays unfavorable electrostatic interactions with Glu 172 and Glu 99. The calculated binding free energies suggest that GLOI prefers the primary enol form (ΔG = − 30.38 kcal/mol) to the keto tautomer (ΔG = − 24.16 kcal/mol). The present work also reveals that bisdemethoxycurcumin binds to GLOI in a similar manner as curcumin and exhibits a slightly less negative predicted binding free energy, which is further validated by our comparative kinetics analysis (Ki = 18.2 and 10.3 μM for bisdemethoxycurcumin and curcumin, respectively). Results of the study can provide an insight into the development of novel and more effective GLOI inhibitors.
Co-reporter:Xuehua Zheng, Liping Zhang, Jing Zhai, Yunyun Chen, ... Xiaopeng Hu
FEBS Letters (2 January 2012) Volume 586(Issue 1) pp:55-59
Publication Date(Web):2 January 2012
DOI:10.1016/j.febslet.2011.11.023
Sulindac (SLD) exhibits both the highest inhibitory activity towards human aldose reductase (AR) among popular non-steroidal anti-inflammatory drugs and clear beneficial clinical effects on Type 2 diabetes. However, the molecular basis for these properties is unclear. Here, we report that SLD and its pharmacologically active/inactive metabolites, SLD sulfide and SLD sulfone, are equally effective as un-competitive inhibitors of AR in vitro. Crystallographic analysis reveals that π–π stacking favored by the distinct scaffold of SLDs is pivotal to their high AR inhibitory activities. These results also suggest that SLD sulfone could be a potent lead compound for AR inhibition in vivo.Highlights► Sulindac and its two metabolites equally inhibit aldose reductase in vitro. ► Crystal structures of sulindacs and tolmetin in complex with aldose reductase. ► Sulindac and its two metabolites have the same binding modes with aldose reductase. ► π–π Stacking favored by sulindac and Phe122 is pivotal to its high inhibition.
Co-reporter:Liping Zhang, Hong Zhang, Yining Zhao, Zhe Li, ... Xiaopeng Hu
FEBS Letters (15 November 2013) Volume 587(Issue 22) pp:3681-3686
Publication Date(Web):15 November 2013
DOI:10.1016/j.febslet.2013.09.031
•Crystal structures of AKR1B10 holoenzyme.•Trp112 side-chain flipping in AKR1B10 explains non-selectivity of AKR1B1 inhibitors.•Native Trp112 side-chain orientation is critical for selective AKR1B10 inhibitors.The antineoplastic target aldo–keto reductase family member 1B10 (AKR1B10) and the critical polyol pathway enzyme aldose reductase (AKR1B1) share high structural similarity. Crystal structures reported here reveal a surprising Trp112 native conformation stabilized by a specific Gln114-centered hydrogen bond network in the AKR1B10 holoenzyme, and suggest that AKR1B1 inhibitors could retain their binding affinities toward AKR1B10 by inducing Trp112 flip to result in an “AKR1B1-like” active site in AKR1B10, while selective AKR1B10 inhibitors can take advantage of the broader active site of AKR1B10 provided by the native Trp112 side-chain orientation.

![(10R)-3c.6t-Dihydroxy-10r.13c-dimethyl-17c-((R)-1.5-dimethyl-hexyl)-(8cH.9tH.14tH)-Delta4-tetradecahydro-1H-cyclopenta[a]phenanthren](http://img.cochemist.com/ccimg/15100/15013-60-2.png)
![(10R)-3c.6t-Dihydroxy-10r.13c-dimethyl-17c-((R)-1.5-dimethyl-hexyl)-(8cH.9tH.14tH)-Delta4-tetradecahydro-1H-cyclopenta[a]phenanthren](http://img.cochemist.com/ccimg/15100/15013-60-2_b.png)

















![(3S,5R,6R,8S,9S,10R,13S,14S)-10,13-Dimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,5,6-triol](http://img.cochemist.com/ccimg/4800/4725-51-3.png)
![(3S,5R,6R,8S,9S,10R,13S,14S)-10,13-Dimethylhexadecahydro-1H-cyclopenta[a]phenanthrene-3,5,6-triol](http://img.cochemist.com/ccimg/4800/4725-51-3_b.png)