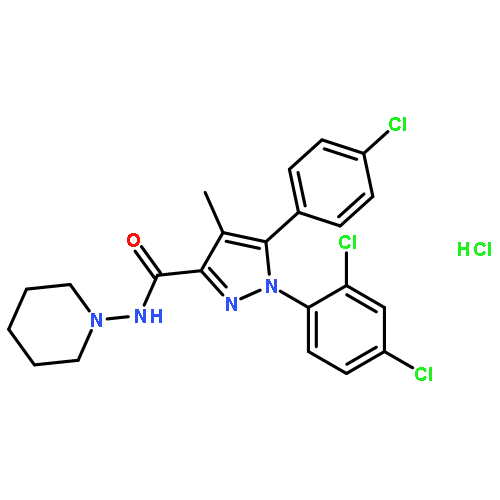
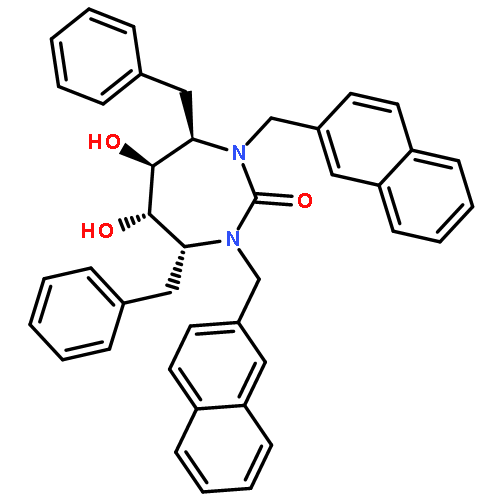
Co-reporter:Zhiye Tang and Chia-en A. Chang
Journal of Chemical Theory and Computation May 9, 2017 Volume 13(Issue 5) pp:2230-2230
Publication Date(Web):April 18, 2017
DOI:10.1021/acs.jctc.6b01204
We introduce a novel method, Pathway Search guided by Internal Motions (PSIM), that efficiently finds molecular dissociation pathways of a ligand–receptor system with guidance from principal component (PC) modes obtained from molecular dynamics (MD) simulations. Modeling ligand–receptor dissociation pathways can provide insights into molecular recognition and has practical applications, including understanding kinetic mechanisms and barriers to binding/unbinding as well as design of drugs with desired kinetic properties. PSIM uses PC modes in multilayer internal coordinates to identify natural molecular motions that guide the search for conformational switches and unbinding pathways. The new multilayer internal coordinates overcome problems with Cartesian and classical internal coordinates that fail to smoothly present dihedral rotation or generate nonphysical distortions. We used HIV-1 protease, which has large-scale flap motions, as an example protein to demonstrate use of the multilayer internal coordinates. We provide examples of algorithms and implementation of PSIM with alanine dipeptide and chemical host–guest systems, 2-naphthyl ethanol−β-cyclodextrin and tetramethylammonium–cryptophane complexes. Tetramethylammonium–cryptophane has slow binding/unbinding kinetics. Its residence time, the length to dissociate tetramethylammonium from the host, is ∼14 s from experiments, and PSIM revealed 4 dissociation pathways in approximately 150 CPU h. We also searched the releasing pathways for the product glyceraldehyde-3-phosphate from tryptophan synthase, and one complete dissociation pathway was constructed after running multiple search iterations in approximately 300 CPU h. With guidance by internal PC modes from MD simulations, the PSIM method has advantages over simulation-based methods to search for dissociation pathways of molecular systems with slow noncovalent kinetic behavior.
Co-reporter:Yu-ming M. HuangMark Anthony V. Raymundo, Wei Chen, Chia-en A. Chang
Biochemistry 2017 Volume 56(Issue 9) pp:
Publication Date(Web):January 6, 2017
DOI:10.1021/acs.biochem.6b01112
Equilibrium constants, together with kinetic rate constants of binding, are key factors in the efficacy and safety of drug compounds, informing drug design. However, the association pathways of protein–ligand binding, which contribute to their kinetic behaviors, are little understood. In this work, we used unbiased all-atom molecular dynamics (MD) simulations with an explicit solvent model to study the association processes of protein–ligand binding. Using the HIV protease (HIVp)–xk263 and HIVp–ritonavir protein–ligand systems as cases, we observed that ligand association is a multistep process involving diffusion, localization, and conformational rearrangements of the protein, ligand, and water molecules. Moreover, these two ligands preferred different routes of binding, which reflect two well-known binding mechanisms: induced-fit and conformation selection models. Our study shows that xk263 has a stronger capacity for desolvating surrounding water molecules, thereby inducing a semiopen conformation of the HIVp flaps (induced-fit model). In contrast, the slow dehydration characteristic of ritonavir allows for gradual association with the binding pocket of HIVp when the protein’s flap conformation is fully open (conformation selection model). By studying the mechanism of ligand association and understanding the role of solvent molecules during the binding event, we can obtain a different perspective on the mechanism of macromolecule recognition, providing insights into drug discovery.
Co-reporter:Christopher C. Roberts and Chia-en A. Chang
The Journal of Physical Chemistry B 2016 Volume 120(Issue 33) pp:8518-8531
Publication Date(Web):June 1, 2016
DOI:10.1021/acs.jpcb.6b02236
We present the second-generation GeomBD Brownian dynamics software for determining interenzyme intermediate transfer rates and substrate association rates in biomolecular complexes. Substrate and intermediate association rates for a series of enzymes or biomolecules can be compared between the freely diffusing disorganized configuration and various colocalized or complexed arrangements for kinetic investigation of enhanced intermediate transfer. In addition, enzyme engineering techniques, such as synthetic protein conjugation, can be computationally modeled and analyzed to better understand changes in substrate association relative to native enzymes. Tools are provided to determine nonspecific ligand–receptor association residence times, and to visualize common sites of nonspecific association of substrates on receptor surfaces. To demonstrate features of the software, interenzyme intermediate substrate transfer rate constants are calculated and compared for all-atom models of DNA origami scaffold-bound bienzyme systems of glucose oxidase and horseradish peroxidase. Also, a DNA conjugated horseradish peroxidase enzyme was analyzed for its propensity to increase substrate association rates and substrate local residence times relative to the unmodified enzyme. We also demonstrate the rapid determination and visualization of common sites of nonspecific ligand–receptor association by using HIV-1 protease and an inhibitor, XK263. GeomBD2 accelerates simulations by precomputing van der Waals potential energy grids and electrostatic potential grid maps, and has a flexible and extensible support for all-atom and coarse-grained force fields. Simulation software is written in C++ and utilizes modern parallelization techniques for potential grid preparation and Brownian dynamics simulation processes. Analysis scripts, written in the Python scripting language, are provided for quantitative simulation analysis. GeomBD2 is applicable to the fields of biophysics, bioengineering, and enzymology in both predictive and explanatory roles.
Co-reporter:Christopher C. Roberts and Chia-en A. Chang
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 1) pp:286-292
Publication Date(Web):December 2, 2014
DOI:10.1021/ct5007482
Colocalized multistep enzymatic reaction pathways within biological catabolic and metabolic processes occur with high yield and specificity. Spatial organization on membranes or surfaces may be associated with increased efficiency of intermediate substrate transfer. Using a new Brownian dynamics package, GeomBD, we explored the geometric features of a surface-anchored enzyme system by parallel coarse-grained Brownian dynamics simulations of substrate diffusion over microsecond (μs) to millisecond (ms) time scales. We focused on a recently developed glucose oxidase (GOx), horseradish peroxidase (HRP), and DNA origami-scaffold enzyme system, where the H2O2 substrate of HRP is produced by GOx. The results revealed and explained a significant advantage in catalytic enhancement by optimizing interenzyme distance and orientation in the presence of the scaffold model. The planar scaffold colocalized the enzymes and provided a diffusive barrier that enhanced substrate transfer probability, becoming more relevant with increasing interenzyme distance. The results highlight the importance of protein geometry in the proper assessment of distance and orientation dependence on the probability of substrate transfer. They shed light on strategies for engineering multienzyme complexes and further investigation of enhanced catalytic efficiency for substrate diffusion between membrane-anchoring proteins.
Co-reporter:Shang Zeng, Yu-ming M. Huang, Chia-en A. Chang and Wenwan Zhong
Analyst 2014 vol. 139(Issue 6) pp:1364-1371
Publication Date(Web):03 Jan 2014
DOI:10.1039/C3AN02155F
Protein adsorption on nanoparticles is closely associated with the physicochemical properties of particles, in particular, their surface properties. We synthesized two batches of polyacrylic acid-coated nanoparticles under almost identical conditions except for the heating duration and found differences in the head-group structure of the polyacrylic acid. The structure change was confirmed by NMR and MS. The two batches of particles had varied binding affinities to a selected group of proteins. Computational work confirmed that the head group of the polymer on the surface of a nanoparticle could directly interact with a protein, and small structural changes in the head group were sufficient to result in a significant difference in the free energy of binding. Our results demonstrate that protein adsorption is so sensitive to the surface properties of particles that it can reveal even small variations in the structure of a nanoparticle surface ligand, and should be useful for quick assessment of nanoparticle properties.
Co-reporter:Yu-ming M. Huang;Myungshim Kang
Journal of Molecular Recognition 2014 Volume 27( Issue 9) pp:537-548
Publication Date(Web):
DOI:10.1002/jmr.2377
Revealing the processes of ligand–protein associations deepens our understanding of molecular recognition and binding kinetics. Hydrogen bonds (H-bonds) play a crucial role in optimizing ligand–protein interactions and ligand specificity. In addition to the formation of stable H-bonds in the final bound state, the formation of transient H-bonds during binding processes contributes binding kinetics that define a ligand as a fast or slow binder, which also affects drug action. However, the effect of forming the transient H-bonds on the kinetic properties is little understood. Guided by results from coarse-grained Brownian dynamics simulations, we used classical molecular dynamics simulations in an implicit solvent model and accelerated molecular dynamics simulations in explicit waters to show that the position and distribution of the H-bond donor or acceptor of a drug result in switching intermolecular and intramolecular H-bond pairs during ligand recognition processes. We studied two major types of HIV-1 protease ligands: a fast binder, xk263, and a slow binder, ritonavir. The slow association rate in ritonavir can be attributed to increased flexibility of ritonavir, which yields multistep transitions and stepwise entering patterns and the formation and breaking of complex H-bond pairs during the binding process. This model suggests the importance of conversions of spatiotemporal H-bonds during the association of ligands and proteins, which helps in designing inhibitors with preferred binding kinetics. Copyright © 2014 John Wiley & Sons, Ltd.
Co-reporter:Zhiye Tang ; Qiao Zhang ; Yadong Yin
The Journal of Physical Chemistry C 2014 Volume 118(Issue 37) pp:21589-21598
Publication Date(Web):August 25, 2014
DOI:10.1021/jp503319s
Colloidal nanomaterials with well-defined shapes have wide applications in many fields. However, the exact role of capping ligands, which often dictates the shape of products in colloidal syntheses, is often unclear. Here we use a classical molecular-mechanics force-field method, mining minima (M2), to compute the binding free energy of the ligands such as citrate, monocarboxylates, dicarboxylates, and tricarboxylates to both (111) and (100) facets of silver and to investigate the mechanisms of the anisotropic growth of silver nanoplates. The distribution of partial charges on a ligand, the geometry complementation in the complex, and the entropic penalty on binding played crucial roles in discriminating the two facets and determining a good or poor ligand. Our finding allows rational design of capping ligands that may perform as well as citrate in promoting the anisotropic growth of nanoplates; however, designing a compound that outperforms citrate is found to be challenging.
Co-reporter:Christopher C. Roberts and Chia-en A. Chang
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 4) pp:2010-2019
Publication Date(Web):March 5, 2013
DOI:10.1021/ct301023m
Modeling binding pathways can provide insight into molecular recognition, including kinetic mechanisms, barriers to binding, and gating effects. This work represents a novel computational approach, Hopping Minima, for the determination of conformational transitions of single molecules as well as binding pathways for molecular complexes. The method begins by thoroughly sampling a set of conformational minima for a molecular system. The natural motions of the system are modeled using the normal modes of the sampled minima. The natural motions are utilized to connect conformational minima and are finally combined to form association/binding pathways in the case of molecular complexes. We provide an implementation and example application of the method using alanine dipeptide and a set of chemical host–guest systems: two cryptophane hosts with two guest cations, trimethylammonium and tetramethylammonium. Our results demonstrate that conformational transitions can be modeled and extended to find binding pathways as well as energetic information relevant to the minimum conformations involved. This approach has advantages over simulation-based methods for studying systems with slow binding processes and can help design molecules with preferred binding kinetics.
Co-reporter:Yu-ming M. Huang, Myungshim Kang, and Chia-en A. Chang
The Journal of Physical Chemistry B 2012 Volume 116(Issue 34) pp:10247-10258
Publication Date(Web):August 3, 2012
DOI:10.1021/jp305028d
Promiscuous proteins are commonly observed in biological systems, for example, in modular domains that recognize phosphopeptides during signal transduction. This promiscuous recognition is of fundamental interest in chemistry and biology but is challenging when designing phosphopeptides in silico for cell biology studies. To investigate promiscuous recognition and binding processes of phosphopeptides and the modular domain, we selected a domain essential in breast cancer—the breast-cancer–associated protein 1 (BRCA1) C-terminal (BRCT) repeats as our model system. We performed molecular dynamics simulations and detailed analyses of the dihedral space to study protein fluctuation and conformational changes with phosphopeptide binding. We also studied the association processes of phosphorylated and unphosphorylated peptides using Brownian dynamics with a coarse-grained model. We found that the BRCT domain is preorganized for phosphopeptide binding but has a moderate arrangement of side chains to form complexes with various types of phosphopeptides. Phosphopeptide binding restricts the system motion in general, while the nonpolar phosphopeptide becomes more flexible in the bound state. Our analysis found that the BRCT domain utilizes different mechanisms, usually termed lock and key, induced-fit, and population-shift/conformational-selection models, to recognize peptides with different features. Brownian dynamics simulations revealed that the charged phosphate group may not always accelerate peptide association processes, but it helps the phosphopeptide orient into binding pockets accurately and stabilizes the complex. This work provides insights into molecular recognition in the promiscuous protein system.
Co-reporter:Rizi Ai, Chia-en A. Chang
Journal of Molecular Graphics and Modelling 2012 Volume 38() pp:155-164
Publication Date(Web):September 2012
DOI:10.1016/j.jmgm.2012.05.002
Cannabinoid (CB1) receptor is a therapeutic drug target, and its structure and conformational changes after ligand binding are of great interest. To study the protein conformations in ligand bound state and assist in drug discovery, CB1 receptor homology models are needed for computer-based ligand screening. The known CB1 ligands are highly diverse structurally, so CB1 receptor may undergo considerable conformational changes to accept different ligands, which is challenging for molecular docking methods. To account for the flexibility of CB1 receptor, we constructed four CB1 receptor models based on four structurally distinct ligands, HU-210, ACEA, WIN55212-2 and SR141716A, using the newest X-ray crystal structures of human β2 adrenergic receptor and adenosine A2A receptor as templates. The conformations of these four CB1–ligand complexes were optimized by molecular dynamics (MD) simulations. The models revealed interactions between CB1 receptor and known binders suggested by experiments and could successfully discriminate known ligands and non-binders in our docking assays. MD simulations were used to study the most flexible ligand, ACEA, in its free and bound states to investigate structural mobility achieved by the rearrangement of the fatty acid chain. Our models may capture important conformational changes of CB1 receptor to help improve accuracy in future CB1 drug screening.Graphical abstractHighlights► Cannabinoid (CB1) receptor models were built with flexible ligand binding sites. ► The models were optimized based on four structurally distinct ligand scaffolds. ► Ligand-induced conformational changes in CB1 receptor were described. ► The models can discriminate known ligands and non-binders in docking assays. ► Our models can help improve accuracy in future CB1 drug screening.
Co-reporter:Myungshim Kang, Christopher Roberts, Yuhui Cheng, and Chia-en A. Chang
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 10) pp:3438-3446
Publication Date(Web):August 26, 2011
DOI:10.1021/ct2004885
Most biological processes are initiated or mediated by the association of ligands and proteins. This work studies multistep, ligand–protein association processes by Brownian dynamics simulations with coarse-grained models for HIV-1 protease (HIVp) and its neutral ligands. We report the average association times when the ligand concentration is 100 μM. The influence of crowding on the simulated binding time was also studied. HIVp has flexible loops that serve as a gate during the ligand binding processes. It is believed that the flaps are partially closed most of the time in its free state. To accelerate our simulations, we fixed a part of the HIVp and reparameterized our coarse-grained model, using atomistic molecular dynamics simulations, to reproduce the “gating” motions of HIVp. HIVp–ligand interactions changed the gating behavior of HIVp and helped ligands diffuse on HIVp surface to accelerate binding. The structural adjustment of the ligand toward its final stable state was the limiting step in the binding processes, which is highly system dependent. The intermolecular attraction between the ligands and crowder proteins contributes the most to the crowding effects. The results highlight broader implications in recognition pathways under more complex environment that considers molecular dynamics and conformational changes. This work brings insights into ligand–protein associations and is helpful in the design of targeted ligands.
Co-reporter:Rizi Ai;M. Qaiser Fatmi
Journal of Computer-Aided Molecular Design 2010 Volume 24( Issue 10) pp:819-827
Publication Date(Web):2010 October
DOI:10.1007/s10822-010-9376-y
T-Analyst is a user-friendly computer program for analyzing trajectories from molecular modeling. Instead of using Cartesian coordinates for protein conformational analysis, T-Analyst is based on internal bond-angle-torsion coordinates in which internal torsion angle movements, such as side-chain rotations, can be easily detected. The program computes entropy and automatically detects and corrects angle periodicity to produce accurate rotameric states of dihedrals. It also clusters multiple conformations and detects dihedral rotations that contribute hinge-like motions. Correlated motions between selected dihedrals can also be observed from the correlation map. T-Analyst focuses on showing changes in protein flexibility between different states and selecting representative protein conformations for molecular docking studies. The program is provided with instructions and full source code in Perl.
Co-reporter:M. Qaiser Fatmi, Rizi Ai and Chia-en A. Chang
Biochemistry 2009 Volume 48(Issue 41) pp:
Publication Date(Web):September 18, 2009
DOI:10.1021/bi901358j
Conformational changes of enzyme complexes are often related to regulating and creating an optimal environment for efficient chemistry. We investigated the synergistic regulation of the tryptophan synthase (TRPS) complex, studied for decades as a model of allosteric regulation and substrate channeling within protein complexes. TRPS is a bifunctional tetrameric αββα enzyme complex that exhibits cooperative motions of the α- and β-subunits by tightly controlled allosteric interactions. We have delineated the atomically detailed dynamics and conformational changes of TRPS in the absence and presence of substrates using molecular dynamics simulations. The computed energy and entropy associated with the protein motions also offer mechanistic insights into the conformational fluctuations and the ligand binding mechanism. The flexible α-L6 loop samples both open and partially closed conformations in the ligand-free state but shifts to fully closed conformations when its substrates are present. The fully closed conformations are induced by favorable protein−ligand interactions but are partly compensated by configurational entropy loss. Considerable local rearrangements exist during ligand binding processes when the system is searching for energy minima. The motion of the region that closes the β-subunit during catalysis, the COMM domain, couples with the motion of the α-subunit, although the fluctuations are smaller than in the flexible loop regions. Because of multiple conformations of ligand-free TRPS in the open and partially closed states, the α-L6 loop fluctuations have preferential directionality, which may facilitate the fully closed conformations induced by α- and β-substrates binding to both subunits. Such cooperative and directional motion may be a general feature that contributes to catalysis in many enzyme complexes.
Co-reporter:Wei Wang;Riccardo Baron;J. Andrew McCammon;William A. McLaughlin
PNAS 2008 Volume 105 (Issue 21 ) pp:7456-7461
Publication Date(Web):2008-05-27
DOI:10.1073/pnas.0800452105
The mechanisms by which a promiscuous protein can strongly interact with several different proteins using the same binding
interface are not completely understood. An example is protein kinase A (PKA), which uses a single face on its docking/dimerization
domain to interact with multiple A-kinase anchoring proteins (AKAP) that localize it to different parts of the cell. In the
current study, the configurational entropy contributions to the binding between the AKAP protein HT31 with the D/D domain
of RII α-regulatory subunit of PKA were examined. The results show that the majority of configurational entropy loss for the
interaction was due to decreased fluctuations within rotamer states of the side chains. The result is in contrast to the widely
held approximation that the decrease in the number of rotamer states available to the side chains forms the major component.
Further analysis showed that there was a direct linear relationship between total configurational entropy and the number of
favorable, alternative contacts available within hydrophobic environments. The hydrophobic binding pocket of the D/D domain
provides alternative contact points for the side chains of AKAP peptides that allow them to adopt different binding conformations.
The increase in binding conformations provides an increase in binding entropy and hence binding affinity. We infer that a
general strategy for a promiscuous protein is to provide alternative contact points at its interface to increase binding affinity
while the plasticity required for binding to multiple partners is retained. Implications are discussed for understanding and
treating diseases in which promiscuous protein interactions are used.
Co-reporter:Yu-ming M. Huang, Wei Chen, Michael J. Potter, Chia-en A. Chang
Biophysical Journal (18 July 2012) Volume 103(Issue 2) pp:
Publication Date(Web):18 July 2012
DOI:10.1016/j.bpj.2012.05.046
Accurate free-energy calculations provide mechanistic insights into molecular recognition and conformational equilibrium. In this work, we performed free-energy calculations to study the thermodynamic properties of different states of molecular systems in their equilibrium basin, and obtained accurate absolute binding free-energy calculations for protein-ligand binding using a newly developed M2 algorithm. We used a range of Asp-Phe-Gly (DFG)-in/out p38α mitogen-activated protein kinase inhibitors as our test cases. We also focused on the flexible DFG motif, which is closely connected to kinase activation and inhibitor binding. Our calculations explain the coexistence of DFG-in and DFG-out states of the loop and reveal different components (e.g., configurational entropy and enthalpy) that stabilize the apo p38α conformations. To study novel ligand-binding modes and the key driving forces behind them, we computed the absolute binding free energies of 30 p38α inhibitors, including analogs with unavailable experimental structures. The calculations revealed multiple stable, complex conformations and changes in p38α and inhibitor conformations, as well as balance in several energetic terms and configurational entropy loss. The results provide relevant physics that can aid in designing inhibitors and understanding protein conformational equilibrium. Our approach is fast for use with proteins that contain flexible regions for structure-based drug design.

![Urea,N-[4-chloro-3-[(3-pyridinyloxy)methyl]phenyl]-N'-[5-(1,1-dimethylethyl)-1H-pyrazol-3-yl]-](http://img.cochemist.com/ccimg/642100/642085-03-8.png)
![Urea,N-[4-chloro-3-[(3-pyridinyloxy)methyl]phenyl]-N'-[5-(1,1-dimethylethyl)-1H-pyrazol-3-yl]-](http://img.cochemist.com/ccimg/642100/642085-03-8_b.png)
![Pyrido[3,2-d]pyrimidin-2(1H)-one, 1-(2,6-dichlorophenyl)-6-[(2,4-difluorophenyl)thio]-3,4-dihydro-7-(1,2,3,6-tetrahydro-4-pyridinyl)-](/data/chemimg/1881600/616894-42-9.png)
![Pyrido[3,2-d]pyrimidin-2(1H)-one, 1-(2,6-dichlorophenyl)-6-[(2,4-difluorophenyl)thio]-3,4-dihydro-7-(1,2,3,6-tetrahydro-4-pyridinyl)-](/data/chemimg/1881600/616894-42-9_b.png)
![S-[(4-formyl-5-hydroxy-6-methylpyridin-3-yl)methyl] dihydrogen phosphorothioate](http://img.cochemist.com/ccimg/69300/69245-32-5.png)
![S-[(4-formyl-5-hydroxy-6-methylpyridin-3-yl)methyl] dihydrogen phosphorothioate](http://img.cochemist.com/ccimg/69300/69245-32-5_b.png)






![Benzamide,3-fluoro-5-(4-morpholinyl)-N-[1-[2-(4-pyridinyl)ethyl]-1H-indol-6-yl]-](http://img.cochemist.com/ccimg/616300/616243-14-2.png)
![Benzamide,3-fluoro-5-(4-morpholinyl)-N-[1-[2-(4-pyridinyl)ethyl]-1H-indol-6-yl]-](http://img.cochemist.com/ccimg/616300/616243-14-2_b.png)
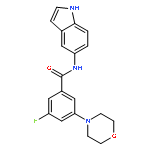
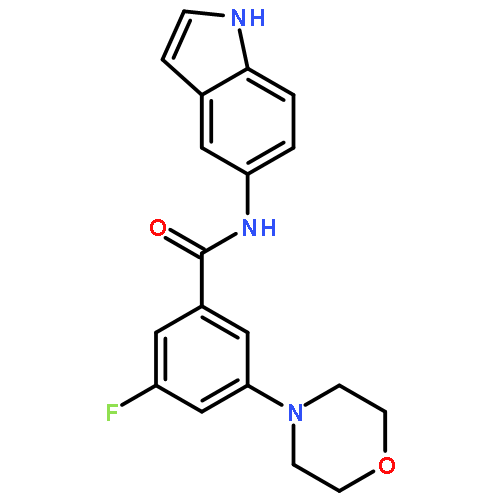
![Benzamide,3-fluoro-5-(4-morpholinyl)-N-[3-[2-(4-pyridinyl)ethyl]-1H-indol-5-yl]-](http://img.cochemist.com/ccimg/616300/616242-87-6.png)
![Benzamide,3-fluoro-5-(4-morpholinyl)-N-[3-[2-(4-pyridinyl)ethyl]-1H-indol-5-yl]-](http://img.cochemist.com/ccimg/616300/616242-87-6_b.png)




![4-Quinazolinamine, 6,7-dimethoxy-N-[3-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/256600/256521-38-7.png)
![4-Quinazolinamine, 6,7-dimethoxy-N-[3-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/256600/256521-38-7_b.png)
![4-[1-(cyclopropylmethyl)-4-(4-fluorophenyl)-1H-imidazol-5-yl]pyrimidin-2-amine](http://img.cochemist.com/ccimg/165900/165806-51-9.png)
![4-[1-(cyclopropylmethyl)-4-(4-fluorophenyl)-1H-imidazol-5-yl]pyrimidin-2-amine](http://img.cochemist.com/ccimg/165900/165806-51-9_b.png)
![Pyridine, 4-[1-(cyclopropylmethyl)-4-(4-fluorophenyl)-1H-imidazol-5-yl]-](http://img.cochemist.com/ccimg/165900/165806-34-8.png)
![Pyridine, 4-[1-(cyclopropylmethyl)-4-(4-fluorophenyl)-1H-imidazol-5-yl]-](http://img.cochemist.com/ccimg/165900/165806-34-8_b.png)
![4-(4-Fluorophenyl)-2-[4-(methylsulfinyl)phenyl]-5-(4-pyridyl)-1H-imidazole](http://img.cochemist.com/ccimg/152200/152121-47-6.png)
![4-(4-Fluorophenyl)-2-[4-(methylsulfinyl)phenyl]-5-(4-pyridyl)-1H-imidazole](http://img.cochemist.com/ccimg/152200/152121-47-6_b.png)


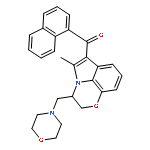
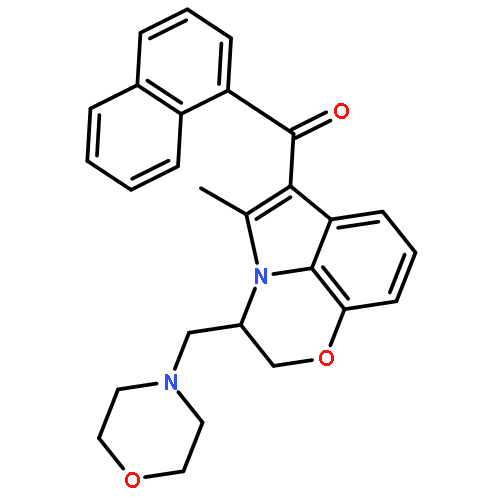




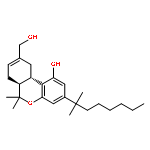
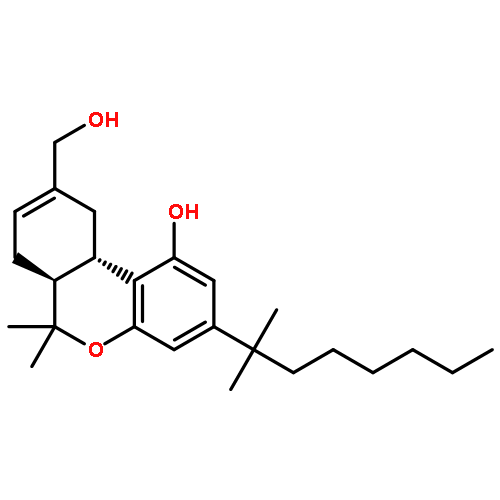


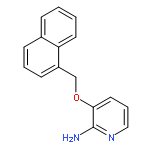
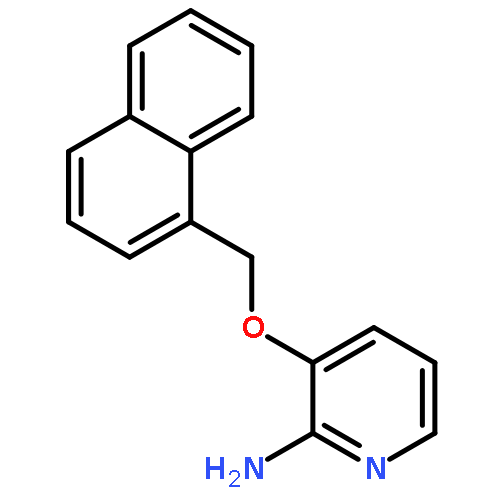
![2-Pyridinamine, 3-[(2,6-dichlorophenyl)methoxy]-](http://img.cochemist.com/ccimg/107300/107229-64-1.png)
![2-Pyridinamine, 3-[(2,6-dichlorophenyl)methoxy]-](http://img.cochemist.com/ccimg/107300/107229-64-1_b.png)




![3-[2-(4-pyridinyl)ethyl]-1H-Indole](http://img.cochemist.com/ccimg/16600/16571-49-6.png)
![3-[2-(4-pyridinyl)ethyl]-1H-Indole](http://img.cochemist.com/ccimg/16600/16571-49-6_b.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)