Co-reporter:Günter Schwarz
BIOspektrum 2017 Volume 23( Issue 1) pp:18-21
Publication Date(Web):2017 February
DOI:10.1007/s12268-017-0759-7
Four molybdenum(Mo)-dependent enzymes are known in humans, each harboring a pterin-based Mo cofactor (Moco), which is synthesized by a conserved biosynthetic pathway. A deficiency in Moco results in a lethal neurodegenerative disorder, for which a therapy has been developed. Moco biosynthesis and enzymes present the origin of various cellular functions ranging from synaptogenesis to nitric oxide synthesis. Understanding this complex interplay aims to translate into new diagnostics and therapies.
Co-reporter:Guenter Schwarz
Current Opinion in Chemical Biology 2016 Volume 31() pp:179-187
Publication Date(Web):April 2016
DOI:10.1016/j.cbpa.2016.03.016
•Molybdenum cofactor is required in two mitochondrial and two cytosolic human enzymes.•Molybdenum enzymes are able to reduce nitrite to nitric oxide (NO).•Deficiency of molybdenum cofactor impairs brain function due to sulfite accumulation.•Sulfite-related changes impact cysteine metabolism provide links to other diseases.•Established and future treatment options for Moco deficiency are discussed.Four molybdenum-dependent enzymes are known in humans, each harboring a pterin-based molybdenum cofactor (Moco) in the active site. They catalyze redox reactions using water as oxygen acceptor or donator. Moco is synthesized by a conserved biosynthetic pathway. Moco deficiency results in a severe inborn error of metabolism causing often early childhood death. Disease-causing symptoms mainly go back to the lack of sulfite oxidase (SO) activity, an enzyme in cysteine catabolism. Besides their name-giving functions, Mo-enzymes have been recognized to catalyze novel reactions, including the reduction of nitrite to nitric oxide. In this review we cover the biosynthesis of Moco, key features of Moco-enzymes and focus on their deficiency. Underlying disease mechanisms as well as treatment options will be discussed.
Co-reporter:Jose Angel Santamaria-Araujo;Victor Wray
JBIC Journal of Biological Inorganic Chemistry 2016 Volume 21( Issue 2) pp:293
Publication Date(Web):2016 April
DOI:10.1007/s00775-015-1321-z
Co-reporter:M Fröhlich, B Dejanovic, H Kashkar, G Schwarz and S Nussberger
Cell Death & Disease 2014 5(2) pp:e1057
Publication Date(Web):2014-02-01
DOI:10.1038/cddis.2014.17
The intrinsic pathway of apoptotic cell death is mainly mediated by the BCL-2-associated X (BAX) protein through permeabilization of the mitochondrial outer membrane (MOM) and the concomitant release of cytochrome c into the cytosol. In healthy, non-apoptotic cells, BAX is predominantly localized in the cytosol and exhibits a dynamic shuttle cycle between the cytosol and the mitochondria. Thus, the initial association with mitochondria represents a critical regulatory step enabling BAX to insert into MOMs, promoting the release of cytochrome c and ultimately resulting in apoptosis. However, the molecular mode of how BAX associates with MOMs and whether a cellular regulatory mechanism governs this process is poorly understood. Here we show that in both primary tissues and cultured cells, the association with MOMs and the proapoptotic action of BAX is controlled by its S-palmitoylation at Cys-126. A lack of BAX palmitoylation reduced BAX mitochondrial translocation, BAX oligomerization, caspase activity and apoptosis. Furthermore, ectopic expression of specific palmitoyl transferases in cultured healthy cells increases BAX S-palmitoylation and accelerates apoptosis, whereas malignant tumor cells show reduced BAX S-palmitoylation consistent with their reduced BAX-mediated proapoptotic activity. Our findings suggest that S-palmitoylation of BAX at Cys126 is a key regulatory process of BAX-mediated apoptosis.
Co-reporter:Jose Angel Santamaria-Araujo;Victor Wray
JBIC Journal of Biological Inorganic Chemistry 2012 Volume 17( Issue 1) pp:113-122
Publication Date(Web):2012 January
DOI:10.1007/s00775-011-0835-2
Hydrogenated (reduced) pterins are found in all living organisms, where they are involved in key metabolic processes. Molybdenum in its biologically active form is bound to a fully reduced tetrahydropyranopterin referred to as a metal-binding pterin (MPT), forming the so-called molybdenum cofactor (Moco). Cyclic pyranopterin monophosphate (cPMP) is the first isolatable intermediate in molybdenum cofactor biosynthesis. Here we present for the first time a 13C NMR characterization of an active Moco intermediate. The 13C NMR data for cPMP corroborate previous data showing the tetrahydropyranopterin nature of cPMP and the presence of a gem-diol in the C1′ position of the side chain. The stability of the gem-diol, together with the absence of any observable signal at low field (175–220 ppm), is an indication that the gem-diol is not a chemical artifact, but is chemically stable and not in equilibrium with the keto form. Finally, we have studied spectrophotometrically the kinetics of cPMP oxidation in the presence of metal centers, chelating agents, and different buffers and pH values. We found that oxidation is metal-dependent and can be substantially retarded in the presence of EDTA.
Co-reporter:Ralf R. Mendel, Günter Schwarz
Coordination Chemistry Reviews 2011 Volume 255(9–10) pp:1145-1158
Publication Date(Web):May 2011
DOI:10.1016/j.ccr.2011.01.054
The transition element molybdenum (Mo) needs to be complexed by a special cofactor in order to gain catalytic activity. With the exception of bacterial Mo-nitrogenase, where Mo is a constituent of the FeMo-cofactor, Mo is bound to a pterin, thus forming the molybdenum cofactor Moco, which in different variants is the active compound at the catalytic site of all other Mo-containing enzymes. The biosynthesis of Moco involves the complex interaction of six proteins and is a process of four steps, which also requires reducing equivalents, iron, ATP and probably copper. After its synthesis, Moco is distributed to the apoproteins of Mo-enzymes by Moco–carrier/binding proteins that also participate in Moco-insertion into the cognate apoproteins. A deficiency in the biosynthesis of Moco has lethal consequences for the respective organisms. In humans, Moco deficiency is a severe inherited inborn error in metabolism resulting in severe neurodegeneration in newborns and causing early childhood death. Due to our better understanding of the chemistry of Moco synthesis, a first therapy has been brought to the clinic.Graphical abstractResearch highlights► Biosynthesis of the pterin-based molybdenum cofactor involves four steps. ► Six proteins catalyze pyranopterin synthesis, sulfur transfer and metal insertion. ► Iron, S-adenosyl methionine, ATP, cysteine and probably copper are needed. ► Moco is distributed by specific carrier/binding proteins to apo-enzymes. ► Moco deficiency is a severe neurodegenerative disorder; a first therapy is available.
Co-reporter:Iris Lambeck, Jen-Chih Chi, Sabina Krizowski, Stefan Mueller, Norbert Mehlmer, Markus Teige, Katrin Fischer, and Guenter Schwarz
Biochemistry 2010 Volume 49(Issue 37) pp:
Publication Date(Web):August 6, 2010
DOI:10.1021/bi1003487
Eukaryotic assimilatory nitrate reductase (NR) is a dimeric multidomain molybdo-heme-flavo protein that catalyzes the first and rate-limiting step in the nitrate assimilation of plants, algae, and fungi. Nitrate reduction takes place at the N-terminal molybdenum cofactor-containing domain. Reducing equivalents are derived from NADH, which reduce the C-terminal FAD domain followed by single-electron transfer steps via the middle heme domain to the molybdenum center. In plants, nitrate reduction is post-translationally inhibited by phosphorylation and subsequent binding of 14-3-3 protein to a conserved phosphoserine located in the surface-exposed hinge between the catalytic and heme domain. Here we investigated Arabidopsis thaliana NR activity upon phosphorylation and 14-3-3 binding by using a fully defined in vitro system with purified proteins. We demonstrate that among different calcium-dependent protein kinases (CPKs), CPK-17 efficiently phosphorylates Ser534 in NR. Out of eight purified Arabidopsis 14-3-3 proteins, isoforms ω, κ, and λ exhibited the strongest inhibition of NR. The kinetic parameters of noninhibited, phosphorylated NR (pNR) and pNR in a complex with 14-3-3 were investigated. An 18-fold reduction in kcat and a decrease in the apparent KMnitrate (from 280 to 141 μM) were observed upon binding of 14-3-3 to pNR, suggesting a noncompetitive inhibition with a preferential binding to the substrate-bound state of the enzyme. Recording partial activities of NR demonstrated that the transfer of electrons to the heme is not affected by 14-3-3 binding. The Ser534Ala variant of NR was not inhibited by 14-3-3 proteins. We propose that 14-3-3 binding to Ser534 blocks the transfer of electrons from heme to nitrate by arresting the domain movement via hinge 1.
Co-reporter:Borislav Dejanovic, Dennis Lal, Claudia B. Catarino, Sita Arjune, Abdel A. Belaidi, Holger Trucks, Christian Vollmar, Rainer Surges, Wolfram S. Kunz, Susanne Motameny, Janine Altmüller, Anna Köhler, Bernd A. Neubauer, EPICURE Consortium, Peter Nürnberg, Soheyl Noachtar, Günter Schwarz, Thomas Sander
Neurobiology of Disease (July 2014) Volume 67() pp:88-96
Publication Date(Web):1 July 2014
DOI:10.1016/j.nbd.2014.02.001
•Identification of novel epilepsy-associated exonic (Δ2–3 and Δ5–9) GPHN deletions•Δ5–9 GPHN impairs oligomerization and clustering of gephyrin at GABAergic synapses.•Δ2–3 GPHN leads to a premature stop-codon after exon 1.•Clinically, intra-cortical inhibition was reduced in the Δ5–9 GPHN patient.•GPHN deletions represent a rare genetic risk factor for IGE.Gephyrin is a postsynaptic scaffolding protein, essential for the clustering of glycine and γ-aminobutyric acid type-A receptors (GABAARs) at inhibitory synapses. An impairment of GABAergic synaptic inhibition represents a key pathway of epileptogenesis. Recently, exonic microdeletions in the gephyrin (GPHN) gene have been associated with neurodevelopmental disorders including autism spectrum disorder, schizophrenia and epileptic seizures. Here we report the identification of novel exonic GPHN microdeletions in two patients with idiopathic generalized epilepsy (IGE), representing the most common group of genetically determined epilepsies. The identified GPHN microdeletions involve exons 5–9 (Δ5–9) and 2–3 (Δ2–3), both affecting the gephyrin G-domain. Molecular characterization of the GPHN Δ5–9 variant demonstrated that it perturbs the clustering of regular gephyrin at inhibitory synapses in cultured mouse hippocampal neurons in a dominant-negative manner, resulting in a significant loss of γ2-subunit containing GABAARs. GPHN Δ2–3 causes a frameshift resulting in a premature stop codon (p.V22Gfs*7) leading to haplo-insufficiency of the gene. Our results demonstrate that structural exonic microdeletions affecting the GPHN gene constitute a rare genetic risk factor for IGE and other neuropsychiatric disorders by an impairment of the GABAergic inhibitory synaptic transmission.
Co-reporter:Matthew Edwards, Juliane Roeper, Catherine Allgood, Raymond Chin, Jose Santamaria, Flora Wong, Guenter Schwarz, John Whitehall
Meta Gene (February 2015) Volume 3() pp:43-49
Publication Date(Web):1 February 2015
DOI:10.1016/j.mgene.2014.12.003
•Molybdenum cofactor deficiency is a severe autosomal recessive metabolic disease.•In neonates it can resemble hypoxemic ischemic encephalopathy.•An affected neonate had high urine L-sulfo-S-cysteine, xanthine, and low blood uric acid.•Compound Z was detected in urine.•Of 2 mutations found in the MOC2A gene, one was shown to disrupt protein expression.BackgroundMolybdenum cofactor deficiency (MOCD) is a severe autosomal recessive neonatal metabolic disease that causes seizures and death or severe brain damage. Symptoms, signs and cerebral images can resemble those attributed to intrapartum hypoxia. In humans, molybdenum cofactor (MOCO) has been found to participate in four metabolic reactions: aldehyde dehydrogenase (or oxidase), xanthine oxidoreductase (or oxidase) and sulfite oxidase, and some of the components of molybdenum cofactor synthesis participate in amidoxime reductase. A newborn girl developed refractory seizures, opisthotonus, exaggerated startle reflexes and vomiting on the second day of life. Treatment included intravenous fluid, glucose supplementation, empiric antibiotic therapy and anticonvulsant medication. Her encephalopathy progressed, and she was given palliative care and died aged 1 week. There were no dysmorphic features, including ectopia lentis but ultrasonography revealed a thin corpus callosum.ObjectivesThe aim of this study is to provide etiology, prognosis and genetic counseling.MethodsBiochemical analysis of urine, blood, Sanger sequencing of leukocyte DNA, and analysis of the effect of the mutation on protein expression.ResultsUric acid level was low in blood, and S-sulfo-L-cysteine and xanthine were elevated in urine. Compound Z was detected in urine. Two MOCS2 gene mutations were identified: c.501 + 2delT, which disrupts a conserved splice site sequence, and c.419C > T (pS140F). Protein expression studies confirmed that the p.S140F substitution was pathogenic. The parents were shown to be heterozygous carriers.ConclusionsMutation analysis confirmed that the MOCD in this family could not be treated with cPMP infusion, and enabled prenatal diagnosis and termination of a subsequent affected pregnancy.



![Thieno[3,2-g]pteridin-4(1H)-one,2-amino-7-[(1R)-1,2-dihydroxyethyl]-6-(methylthio)- (9CI)](http://img.cochemist.com/ccimg/19300/19295-31-9.png)
![Thieno[3,2-g]pteridin-4(1H)-one,2-amino-7-[(1R)-1,2-dihydroxyethyl]-6-(methylthio)- (9CI)](http://img.cochemist.com/ccimg/19300/19295-31-9_b.png)














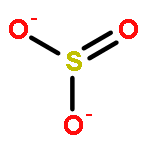
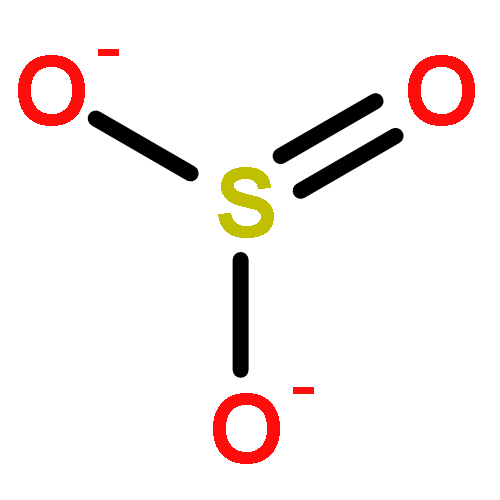


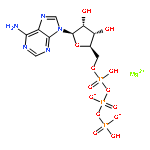



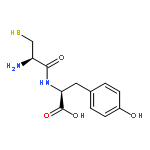




![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)

