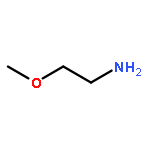
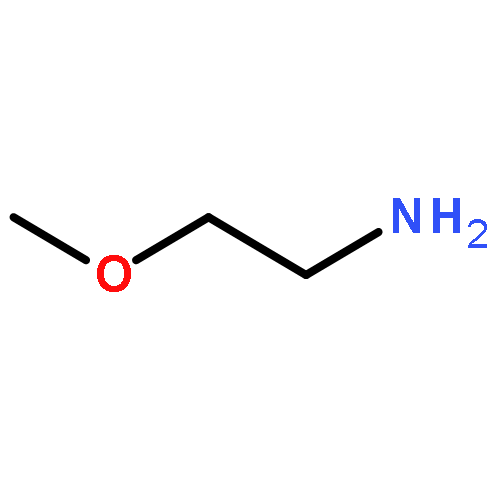
Co-reporter:Guanyi Wang 王冠祎;Wantong Song 宋万通;Na Shen 沈娜;Haiyang Yu 于海洋
Science China Materials 2017 Volume 60( Issue 10) pp:995-1007
Publication Date(Web):27 September 2017
DOI:10.1007/s40843-017-9107-x
Osteosarcoma is a high-class malignant bone cancer with a less than 20% five-year survival rate due to its early metastasis potential. There is an urgent need to develop a versatile and innoxious drug to treat metastatic osteosarcoma. Curcumin (Cur) has shown its potential for the treatment of many cancers; however, the clinical implication of native curcumin is severely hindered by its intrinsic property. In this study, a mixed system of monomethoxy (polyethylene glycol)-poly(d, l-lactide-co-glycolide)/poly(ε-caprolactone) (mPEG-PLGA/PCL) was used to build a formulation of curcumin-encapsulated nanoparticles (Cur-NPs), which significantly improved the solubility, stability and cellular uptake of curcumin. Moreover, the Cur-NPs were superior to free curcumin in the matter of inhibition on the proliferation, migration and invasion of osteosarcoma 143B cells. It was found that both free curcumin and Cur-NPs could decrease the expressions of c-Myc and MMP7 in the level of mRNA and protein, which explained why free curcumin and Cur-NPs could inhibit the proliferation and invasion of metastatic osteosarcoma 143B cells. The Cur-NPs provided a promising strategy for metastatic osteosarcoma treatment.骨肉瘤是一种高转移性的恶性肿瘤, 5年生存率不到20%.姜黄素(Cur)具有治疗癌症的潜在功效, 但其自身水溶性和稳定性差等性质限制了其临床使用. 本文用聚乙二醇-聚(D, L-丙交酷-乙交酷)/聚(ε-已内酷)负载姜黄素形成纳米粒子, 可以改善姜黄素的水溶性、 稳定性和细胞内吞. 与非负载的姜黄素相比, 姜黄素纳米粒子在抑制骨肉瘤细胞增殖和迁移方面显示出明显的优势. 研究结果表明, 姜黄素和其纳米粒子均可降低c-Myc和MMP7表达, 这可以很好地解释姜黄素纳米粒子抑制骨肉瘤细胞的机制.
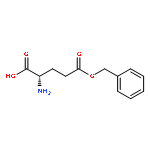
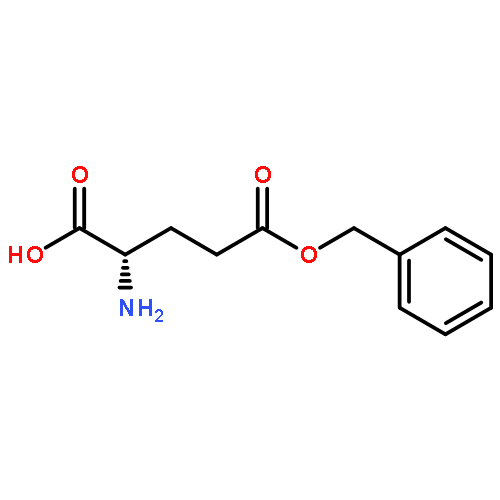
A pH and redox dual-sensitive biodegradable polysaccharide, succinic acid-decorated dextran-g-phenylalanine ethyl ester-g-cysteine ethyl ester (Dex-SA-L-Phe-L-Cys), was synthesized to load doxorubicin hydrochloride (DOX·HCl). The DOX-loaded nanoparticles, which were prepared in aqueous solution and free of organic solvent, could spontaneously self-assemble into uniform sizes. When loading DOX·HCl, mercapto Dex-SA-L-Phe-L-Cys was oxidized into a crosslinked disulfide linkage to form pH and redox dual-sensitive nanoparticles (DOX-S-NPs). The amphiphilic polymer loaded DOX·HCl into the core through electrostatic and hydrophobic interactions, meanwhile the crosslinked disulfide bond could stabilize the drug loaded nanoparticles. As a control with similar polymer structure, succinic acid decorated dextran-g-phenylalanine ethyl ester (Dex-SA-L-Phe) was prepared to obtain pH-sensitive DOX-loaded micelles (DOX-N-NPs). The controlled pH and redox-dependent release profiles of the DOX-S-NPs in vitro were certified in different releasing mediums. Furthermore, the cellular uptake of the DOX-S-NPs was comparable with that of free DOX·HCl, determined by confocal laser scanning microscopy (CLSM) and flow cytometry. Cytotoxicity assay in vitro showed that the DOX-S-NPs and free DOX·HCl were similar in inhibiting the proliferation of non-small cell lung carcinoma A549 and breast cancer MCF-7 cell lines. DOX-S-NPs displayed similar antitumor efficiency compared with free DOX·HCl, but lower toxicity by body weight. These dual-sensitive DOX-S-NPs provide a useful strategy for anti-tumor therapy.
Combretastatin A4 (CA4) is a leading agent in vascular disrupting strategies for tumor therapy. Although many small-molecule prodrugs of CA4 have been developed to improve its solubility, the overall therapeutic efficiency is moderate. A key reason for this is the reversible effect that CA4 has on tubulin as well as its rapid clearance from plasma and tissues. In this study, we proposed a poly(l-glutamic acid)-CA4 conjugate (PLG-CA4) nanomedicine to fulfill the requirements for fully liberating the potential of CA4 on tumor therapy. Enhanced accumulation and retention of CA4 in tumor tissue, especially, high distribution and gradual release around tumor blood vessels resulted in prolonged vascular disruption and markedly enhanced therapeutic efficiency. We examined and compared the therapeutic effect of PLG-CA4 and commercial combretastatin-A4 phosphate (CA4P) in a murine colon C26 tumor. PLG-CA4 showed significantly prolonged retention in plasma and tumor tissue. Most importantly, the PLG-CA4 was mainly distributed around the tumor vessels because of its low tissue penetration in solid tumor. Pathology tests showed that PLG-CA4 treatment resulted in persistent vascular disruption and tumor damage 72 h after a single injection, this in contrast to CA4P treatment, which showed quick relapse at an equal dose. Tumor suppression tests showed that PLG-CA4 treatment resulted in a tumor suppression rate of 74%, which indicates a significant advantage when compared to tumor suppression rate of the CA4P group, which was 24%. This is the first time that an advantage of the polymeric CA4 nanomedicine with low tissue penetration for solid tumor therapy has been shown. Thus, the results presented in this study provide a new idea for enhancing the tumor therapeutic effect of vascular disrupting agents.Statement of SignificanceNanomedicine usually has low tissue penetration in solid tumors, which limits the efficacy of nanomedicine in most cases. But herein, we demonstrate a nanosized vascular disruptive agent (VDA) PLG-CA4 has supper advantages over small molecular combretastatin-A4 phosphate (CA4P) because the PLG-CA4 was mainly distributed around the tumor vessels due to its low tissue penetration in solid tumor.Download high-res image (214KB)Download full-size image
Traditional chemotherapy strategy exists undesirable toxic side-effects to normal tissues due to the low selectively to cancer cells of micromolecule cytotoxic drugs. One considered method to realizing the targeted delivery and increasing the specificity to tumor tissues of the cytotoxic drug is to transporting and discharging it through an environment-sensitive mechanism. In this study, a novel enzyme-sensitive polymer-doxorubicin conjugate was designed to delivery chemotherapeutic drug in a tumor-specific behavior and selectively activated in tumor tissue. Briefly, doxorubicin (DOX) was conjugated to carboxyl-terminated 4-arm poly(ethylene glycol) through a tetrapeptide linker, alanine-alanine-asparagine-leucine (AANL), which was one of the substrates of legumain, an asparaginyl endopeptidase that was found presented in plants, mammals and also highly expressed in human tumor tissues. Hereinafter, the polymer-DOX conjugate was termed as 4-arm PEG-AANL-DOX. Dynamic laser scattering (DLS) and transmission electron microscopy (TEM) measurements indicated that the 4-arm PEG-AANL-DOX could self-assemble into micelles in aqueous solution. Drug release and in vitro cytotoxicity studies revealed that the 4-arm PEG-AANL-DOX could be cleaved by legumain. Ex vivo DOX fluorescence imaging measurements demonstrated that the 4-arm PEG-AANL-DOX had an improved tumor-targeting delivery as compared with the free DOX·HCl. In vivo studies on nude mice bearing MDA-MB-435 tumors revealed that the 4-arm PEG-AANL-DOX had a comparable anticancer efficacy with the free DOX·HCl but without DOX-related toxicities to normal tissues as measured by body weight change and histological assessments, indicating that the 4-arm PEG-AANL-DOX had an improved therapeutic index for cancer therapy.Statement of SignificanceHerein we describe the construction of a novel tumor environment-sensitive delivery system through the instruction of a legumain-cleavable linkage to a polymer-DOX conjugate (4-arm PEG-AANL-DOX). This particular design strategy allows for polymer-DOX conjugates to be delivered in a tumor-specific manner and selectively activable in tumor microenvironment so that it can combine the advantages of tumor-specific delivery and tumor intracellular microenvironment-triggered release systems.Download high-res image (126KB)Download full-size image
In the pursuit of effective treatments for cancer, an emerging strategy is “active targeting”, where nanoparticles are decorated with targeting ligands able to recognize and bind specific receptors overexpressed by tumor cells or tumor vasculature so that a greater fraction of the administered drugs are selectively trafficked to tumor sites. However, the implementation of this strategy has faced a major obstacle. The interpatient, inter- and intra-tumoral heterogeneity in receptor expression can pose challenges for the design of clinical trials and result in the paucity of targetable receptors within a tumor, which limits the effectiveness of “active targeting” strategy in cancer treatment. Here we report a cooperative drug delivery platform that overcomes the heterogeneity barrier unique to solid tumors. The cooperative platform comprises a coagulation-inducing agent and coagulation-targeted polymeric nanoparticles. As a typical small-molecule vascular disrupting agent (VDA), DMXAA can create a unique artificial coagulation environment with additional binding sites in a solid tumor by locally activating a coagulation cascade. Coagulation-targeted cisplatin-loaded nanoparticles, which are surface-decorated with a substrate of activated blood coagulation factor XIII, can selectively accumulate in the solid tumor by homing to the VDA-induced artificial coagulation environment through transglutamination. In vivo studies show that the cooperative tumor-selective platform recruits up to 7.5-fold increases in therapeutic cargos to the tumors and decreases tumor burden with low systemic toxicity as compared with non-cooperative controls. These indicate that the use of coagulation-targeted nanoparticles, in conjunction with free small-molecule VDAs, may be a valuable strategy for improving standard chemotherapy.
Journal of Controlled Release 2016 Volume 231() pp:94-102
Publication Date(Web):10 June 2016
DOI:10.1016/j.jconrel.2016.02.039
A poly(l-glutamic acid) graft polyethylene glycol-cisplatin complex (PGA-CisPt) performs well in reducing the toxicity of free cisplatin and greatly enhances the accumulation and retention of cisplatin in solid tumors. However, there is a lack of effective treatment options for cisplatin-resistant tumors. A major reason for this is the dense PEG shell, which ensures that the PGA-CisPt maintains a long retention time in the blood that may result in it bypassing the tumor cells or failing to be endocytosed within the tumor microenvironment. Consequently, the cisplatin from PGA-CisPt is released to the extracellular space in the presence of cisplatin-resistant tumor cells and the resistant problem to free cisplatin still valid. Therefore, we devised a strategy to combat the resistance of cisplatin in the tumor microenvironment using nanoparticles-loaded disulfiram (NPs-DSF) as a modulator. In vitro, cisplatin, in combination with DSF, had a synergistic effect and decreased cell survival rate of cisplatin-resistant A549DDP cells. This effect was also observed when combining PGA-CisPt with NPs-DSF. Similarly, in Balb/C nude mice with A549DDP xenografts, NPs-DSF improved PGA-CisPt effectiveness in inhibiting tumor growth while maintaining low toxicity. Our data demonstrate that DSF reduces intracellular glutathione (GSH) levels, inhibits NFκB activity, and modulates the expression of apoptosis-related proteins Bcl-2 and Bax, thereby improves the effectiveness of cisplatin in resistant cell lines. Here, we provide a promising method for overcoming cisplatin resistance in tumors, while maintaining the in vivo benefits of the PGA-CisPt complex.
Stimuli-responsive drug-delivery systems have attracted great attention for controlled drug release. The pH value is relatively low in the tumor tissues and the concentration of glutathione is significantly high in the cells. According to these features, a charge-conversional polymeric prodrug that is sensitive to pH and redox is designed. 3,3′-Dithiodipropionic acid modified paclitaxel (DTPA-PTX) and 2,3-dimethylmalefic anhydride (DMMA) are conjugated to the amino groups of poly(ethylene glycol)-b-poly(L-lysine) (mPEG-b-PLL). The surface charge of the obtained mPEG-b-(PLL-co-(PLL-DMMA)-co-(PLL-DTPA-PTX)) nanoparticles (DMMA-NPs) can change from negative at blood pH to positive at tumor extracellular pH, implying that the DMMA-NPs will have a prolonged blood circulation time through avoiding rapid clearance by the reticuloendothelial system and an enhanced cell uptake at the tumor site. Compared to the control group, the in vitro studies show that the DMMA-NPs exhibit effective cellular uptake, rapid intracellular drug release, and significantly enhanced cytotoxicity against MCF-7 tumor cells, owing to the tumor-relevant pH and reductive conditions. These indicate that the charge-conversional DMMA-NPs provide an excellent platform for potential tumor therapy.
Photoacoustic imaging (PAI) is an emerging modality in biomedical imaging. Photoacoustic effect is the basis for PAI, where a photoacoustic contrast agent absorbs optical pulses to initiate localized heating and rapid thermal expansion, thus generating thermoelastic stress waves. Therefore, ideal PAI dyes should have strong NIR light absorbance and high light-heat conversion efficiency. However, most current low molecular weight organic PAI contrast agents are fluorescent dyes, where the light-heat conversion efficiency is dramatically impaired due to the energy loss by fluorescence emission. Herein, we report a series of highly efficient photoacoustic dyes with COOH, NH2 and NHS ester functionalities, from an inexpensive industrial computer-to-plate NIR absorber (IR830 p-toluenesulfonate) that has a strong NIR absorbance but an extremely low fluorescence emission. In vitro and in vivo studies show that the functional IR830 dyes have low cytotoxicity, and are 2.1 folds brighter in photoacoustic imaging than traditional photoacoustic dye indocyanine green (ICG). The Lowest Limit of Quantification of the IR830 series dyes is as low as the 1/7 of that of ICG. These indicate that the functional IR830 dyes have great potential as highly efficient photoacoustic dyes.
Statement of Significance
Photoacoustic imaging (PAI) is an emerging modality in biomedical imaging. Ideal PAI dyes should have strong NIR absorbance and high light-heat conversion efficiency. However, most current low molecular weight organic PAI contrast agents are fluorescent dyes, where the light-heat conversion efficiency is dramatically impaired due to the energy loss by fluorescence emission. Herein we report a series of highly efficient functional photoacoustic dyes from an inexpensive industrial computer-to-plate NIR absorber (IR830) that has a strong NIR absorbance but an extremely low fluorescence emission. The functional IR830 dyes show low cytotoxicity, much brighter in photoacoustic imaging than traditional photoacoustic dye indocyanine green. These indicate that the functional IR830 dyes have great potential as highly efficient photoacoustic dyes.
Nanomedicine: Nanotechnology, Biology and Medicine 2016 Volume 12(Issue 2) pp:377-386
Publication Date(Web):February 2016
DOI:10.1016/j.nano.2015.10.022
Disulfiram (DSF) showed great potential in an in vitro tumor therapy study; however, those results could not be applied to an in vivo study due to the extreme instability of DSF in blood. Here, we describe a system of methoxy poly(ethylene glycol)-b-poly(lactide-co-glycolide)/poly(ε-caprolactone) (mPEG-PLGA/PCL) mixed nanoparticles (NPs) for DSF loading and delivery. By adjusting the mPEG-PLGA/PCL content ratios, the DSF loading capacity increased to 7.8%, while the hydrodynamic radii of the NPs were around 50-100 nm. The DSF-loaded NPs showed high stability in distilled water and 10% serum-containing phosphate buffered saline. The NPs efficiently protected DSF from degradation while maintaining its anti-tumor properties. Furthermore, a pharmacokinetics study demonstrated that NP delivery system enhanced the DSF concentration in the blood after tail vein injection. Finally, DSF delivery using this model effectively slowed the growth of a 4T1 murine xenograft tumor.From the Clinical EditorThe anti-tumor efficacy of the anti-alcoholic drug disulfiram has been known for some time. However, its use in the clinical setting is limited due to the underlying instability of the drug. In this study, the authors utilized a nanocarrier system of mPEG-PLGA/PCL for the delivery of this drug. The promising results may allow encapsulation of other drugs.Disulfiram was loaded inside mPEG-PLGA/PCL mixed nanoparticles by nanoprecipitation, and the formed nanoparticles showed good disulfiram loading capacity and stability, and effectively protect disulfiram from degradation. In vivo studies proved that after entrapment, disulfiram was effective in in vivo tumor inhibition. Thus, the formula applied here made this un-applicable drug into applicable.
Journal of Controlled Release 2015 Volume 205() pp:89-97
Publication Date(Web):10 May 2015
DOI:10.1016/j.jconrel.2014.12.022
Platinum-based polymeric nano-drugs, especially cisplatin-loaded polymeric nanoparticles (CDDP-NPs), have been extensively exploited for the treatment of solid tumors. However, it is still unclear what role the processing procedure and the properties of the polymeric carrier materials may play in influencing the plasma pharmacokinetics, biodistribution and in vivo efficacy of CDDP-NPs. In this study, a series of poly(l-glutamic acid)-g-methoxy poly(ethylene glycol) (PLG-g-mPEG) copolymers were synthesized for the preparation of CDDP-loaded PLG-g-mPEG (CDDP/PLG-g-mPEG) nanoparticles. All of the parameters, including PLG molecular weight, mPEG/PLG weight ratio, mPEG chain length, ultrafiltration purification and cisplatin loading content, were found to have a significant influence on the plasma pharmacokinetics of the CDDP/PLG-g-mPEG nanoparticles. The blood circulation time of the nanoparticles was prolonged with increases in PLG molecular weight, mPEG/PLG weight ratio, mPEG chain length and CDDP loading content. The use of ultrafiltration purification could prolong the blood circulation time of the nanoparticles as well. Experiments to measure the pharmacokinetics and biodistribution demonstrated that the selected CDDP/PLG-g-mPEG nanoparticles, NP10, had a long blood circulation time and could achieve selective and significant accumulation in Lewis lung carcinoma (LLC) tumors. The platinum plasma concentrations in the LLC tumor-bearing mice receiving NP10 remained up to 46-fold higher than that of mice receiving equivalent doses of free CDDP. In addition, the plasma area under the concentration time curve (AUC) of NP10 was 31-fold higher than that of free CDDP in 48 h. The platinum concentration ratio of NP10 to free CDDP in tumors reached as high as 9.4. The tumor AUC ratio of NP10 to CDDP was 6. Using a mouse C26 tumor model, here we demonstrate that NP10 improves the safety and tolerance in vivo when compared to CDDP and effectively inhibits the growth of C26 tumors. Furthermore, increasing the dosage of NP10 by 2 or 3-fold of free CCDP improved its anticancer efficacy to comparable or higher levels. These results indicate that CDDP/PLG-g-mPEG nanoparticles have greater potential for the treatment of solid tumors in clinical application.
Cisplatin-loaded poly(l-glutamic acid)-g-methoxy poly(ethylene glycol 5K) nanoparticles (CDDP-NPs) were characterized and exploited for the treatment of non-small cell lung carcinoma (NSCLC). In vitro metabolism experiments showed that a glutamic acid 5-mPEG ester [CH3O(CH2CH2O)nGlu] was generated when the poly(l-glutamic acid)-g-methoxy poly(ethylene glycol 5K) (PLG-g-mPEG5K) was incubated with HeLa cells. This suggests that the poly(glutamic acid) backbone of the PLG-g-mPEG5K is biodegradable. Furthermore, the size of the CDDP-NPs in an aqueous solution was affected by varying the pH (5.0–8.0) and their degradation rate was dependent on temperature. The CDDP-NPs could also bind to the model nucleotide 2′-deoxyguanosine 5′-monophosphate, indicating a biological activity similar to cisplatin. The CDDP-NPs showed a significantly lower peak renal platinum concentration after a single systemic administration when compared to free cisplatin. In vivo experiments with a Lewis lung carcinoma (LLC) model showed that the CDDP-NPs suppressed the growth of tumors. In addition, LLC tumor-bearing mice treated with the CDDP-NPs (5 mg/kg cisplatin eq.) showed much longer survival rates (median survival time: 51 days) as compared with mice treated with free cisplatin (median survival time: 18 days), due to the acceptable antitumor efficacy and low systemic toxicity of CDDP-NPs. These results suggest that the CDDP-NPs may be successfully applied to the treatment of NSCLC.
A novel random copolypeptide of ornithine, arginine, glycine, and aspartic acid [Poly(ornithine-co-arginine-co-glycine-co-aspartic acid), Poly(O,R,G,D)] has been prepared through ring-opening polymerization of N-δ-carbobenzoxy-l-ornithine N-carboxyanhydride [Orn(Cbz)-NCA)], l-glycine N-carboxyanhydride (Gly-NCA) and β-benzyl l-aspartate N-carboxyanhydride [Asp(Bn)-NCA], following by subsequent deprotection and guanidization. The structure of Poly(O,R,G,D) was confirmed by nuclear magnetic resonance (NMR) spectroscopy and gel permeation chromatography (GPC). Low cytotoxicity of Poly(O,R,G,D) was confirmed from MTT assay. The Poly(O,R,G,D) contain some internal sequences of RXXR (X = O, R, G, or D) that could be proteolytically cleaved to expose the cryptic CendR element and bind to Neuropilin-1. This would lead to vascular and tissue permeabilization. Therefore trypsin-cleaved Poly(O,R,G,D) increase the vascular leakage of Evans blue from dermal microvessels at the injection site in vivo skin permeability assay. The intratumoral injection of the Poly(O,R,G,D) significantly enhanced the concentration of cisplatin-loaded nanoparticles in MCF-7 solid tumors. These results show that Poly(O,R,G,D) could increase the vascular leakage and tissue penetration of nanoparticles in a solid tumor and can be used as a potential polymeric tumor-penetrating agent.
Real-time and continuous monitoring of systemically administered agents is an important task in pharmaceutical development. Herein, we performed a real-time continuous study of the pharmacokinetics and biodistribution of indocyanine green (ICG) and liposomal indocyanine green (Lipo-ICG) in vivo by multispectral optoacoustic tomography (MSOT). By comparing the blood clearance and uptake behavior of these two ICG formulations in liver, spleen, kidney and tumor, we showed that Lipo-ICG prolonged the retention time of ICG in blood, and resulted in enhanced accumulation and retention in liver, spleen, and tumor. The results obtained from the MSOT test provided a comprehensive and continuous view of the metabolic behavior of the injected agents in different formulations. The results may also be helpful for understanding this new imaging technique.
Co-reporter:Yi-fei Li;Hai-yang Yu;Hai Sun;Jian-guo Liu 刘建国
Chinese Journal of Polymer Science 2015 Volume 33( Issue 5) pp:763-771
Publication Date(Web):2015 May
DOI:10.1007/s10118-015-1624-0
Herein, cisplatin-loaded poly(L-glutamic acid)-g-methoxy poly(ethylene glycol) nanoparticles were evaluated as a potential chemotherapeutic agent against osteosarcoma by using alone or with an iRGD (internalizing RGD, CRGDKDPDC). The release rate of platinum from the cisplatin-loaded nanoparticles CDDP/PLG160-g-mPEG2K (CDDP-NPs) accelerated with the increase of the acidity of the environment. In vitro test demonstrated that CDDP-NPs could inhibit the proliferation of MNNG/Hos osteosarcoma cells with IC50 (72 h) of 12.2 μg·mL−1. In vivo test for MNNG/Hos osteosarcoma tumor bearing mice exhibited that CDDP-NPs had comparable or slightly higher efficacy but significantly lower side effects in comparison with free CDDP. The coadministration of iRGD could further enhance the anticancer efficacy of CDDP-NPs against MNNG/Hos osteosarcoma without bringing obvious side effects. Therefore, CDDP-NPs using alone or with iRGD have great potential for the treatment of osteosarcoma.
A novel methoxy poly(ethylene glycol)-b-poly(l-glutamic acid)-b-poly(l-phenylalanine) (mPEG-b-P(Glu)-b-P(Phe)) triblock copolymer was prepared and explored as a micelle carrier for the co-delivery of paclitaxel (PTX) and cisplatin (cis-diamminedichlo-platinum, CDDP). PTX and CDDP were loaded inside the hydrophobic P(Phe) inner core and chelated to the middle P(Glu) shell, respectively, while mPEG provided the outer corona for prolonged circulation. An in vitro release profile of the PTX + CDDP-loaded micelles showed that the CDDP chelation cross-link prevented an initial burst release of PTX. The PTX + CDDP-loaded micelles exhibited a high synergism effect in the inhibition of A549 human lung cancer cell line proliferation over 72 h incubation. For the in vivo treatment of xenograft human lung tumor, the PTX + CDDP-loaded micelles displayed an obvious tumor inhibiting effect with a 83.1% tumor suppression rate (TSR%), which was significantly higher than that of a free drug combination or micelles with a single drug. In addition, more importantly, the enhanced anti-tumor efficacy of the PTX + CDDP-loaded micelles came with reduced side-effects. No obvious body weight loss occurred during the treatment of A549 tumor-bearing mice with the PTX + CDDP-loaded micelles. Thus, the polypeptide-based combination of PTX and CDDP may provide useful guidance for effective and safe cancer chemotherapy.
Cisplatin (cis-diaminodichloroplatinum, CDDP) loaded methoxy poly (ethylene glycol)-block-poly (glutamic acid-co-phenyl alanine) [mPEG-b-P (Glu10-co-Phe10) (PGlu10) and mPEG-b-P (Glu20-co-Phe10) (PGlu20)] nanoparticles with two different formulations (CDDP/PGlu10 and CDDP/PGlu20) are successfully developed in uniformly sizes. In 190 h, the CDDP/PGlu10 shows 30% release at physiological pH and 39% at lysosomal pH. Similarly, the CDDP/PGlu20 shows 60% release at physiological pH and 90% release at lysosomal pH. The sustained and controlled release of both formulations evidences the in vitro longevity of the nanoparticles. The cell proliferation inhibition of nanoparticles against human breast cancer cell line ZR-75-30 is dose and time dependent. Both CDDP/PGlu10 and CDDP/PGlu20 show excellent hemo compatibility as evaluated by hemolysis experiments. The in vivo fate of CDDP and CDDP loaded nanoparticles are evaluated by pharmacokinetics studies. Free CDDP underwgoes instant platinum concentration decrease after intravenous administration with 1.0 wt% left in 24 h while the CDDP loaded nanoparticles show prolonged blood circulation time with 5 wt% (CDDP/PGlu20) to 14 wt% (CDDP/PGlu10) left in 24 h. This prolonged blood circulation of CDDP loaded nanoparticles makes them as promising nanocarriers for tumor targeting delivery.
Amphiphilic linear and dumbbell-shaped poly(ethylene glycol)–poly(lactide-co-glycolide) (PEG–PLGA) copolymers were simply synthesized by the ring-opening polymerization of lactide and glycolide using PEG or tetrahydroxyl-functionalized PEG as the macroinitiator and stannous octoate as the catalyst. The copolymers spontaneously self-assembled into spherical micelles in phosphate-buffered saline at pH 7.4. The self-assembly behavior was dependent on both the polymeric topology and composition. Doxorubicin (DOX), an anthracycline antitumor drug, was loaded into micelles through nanoprecipitation. The in vitro release behavior could be adjusted by regulating the topology or composition of the copolymer, or the pH of the release medium. The effective intracellular DOX release from DOX-loaded micelles was confirmed by confocal laser scanning microscopy and flow cytometry in vitro. DOX-loaded micelles displayed great cellular proliferation inhibition efficacies after incubation for 24, 48 or 72 h. The hemolysis ratio of DOX was significantly reduced by the presence of copolymer. These properties indicated that the micelles derived from linear or dumbbell-shaped copolymers were promising candidates as smart antitumor drug carriers for malignancy therapy.
Chemical Science (2010-Present) 2016 - vol. 7(Issue 1) pp:NaN736-736
Publication Date(Web):2015/10/26
DOI:10.1039/C5SC01698C
In the pursuit of effective treatments for cancer, an emerging strategy is “active targeting”, where nanoparticles are decorated with targeting ligands able to recognize and bind specific receptors overexpressed by tumor cells or tumor vasculature so that a greater fraction of the administered drugs are selectively trafficked to tumor sites. However, the implementation of this strategy has faced a major obstacle. The interpatient, inter- and intra-tumoral heterogeneity in receptor expression can pose challenges for the design of clinical trials and result in the paucity of targetable receptors within a tumor, which limits the effectiveness of “active targeting” strategy in cancer treatment. Here we report a cooperative drug delivery platform that overcomes the heterogeneity barrier unique to solid tumors. The cooperative platform comprises a coagulation-inducing agent and coagulation-targeted polymeric nanoparticles. As a typical small-molecule vascular disrupting agent (VDA), DMXAA can create a unique artificial coagulation environment with additional binding sites in a solid tumor by locally activating a coagulation cascade. Coagulation-targeted cisplatin-loaded nanoparticles, which are surface-decorated with a substrate of activated blood coagulation factor XIII, can selectively accumulate in the solid tumor by homing to the VDA-induced artificial coagulation environment through transglutamination. In vivo studies show that the cooperative tumor-selective platform recruits up to 7.5-fold increases in therapeutic cargos to the tumors and decreases tumor burden with low systemic toxicity as compared with non-cooperative controls. These indicate that the use of coagulation-targeted nanoparticles, in conjunction with free small-molecule VDAs, may be a valuable strategy for improving standard chemotherapy.









![disodium,[2-methoxy-5-[(Z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenyl] phosphate](http://img.cochemist.com/ccimg/168600/168555-66-6.png)
![disodium,[2-methoxy-5-[(Z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenyl] phosphate](http://img.cochemist.com/ccimg/168600/168555-66-6_b.png)
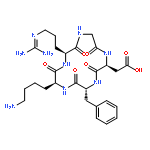
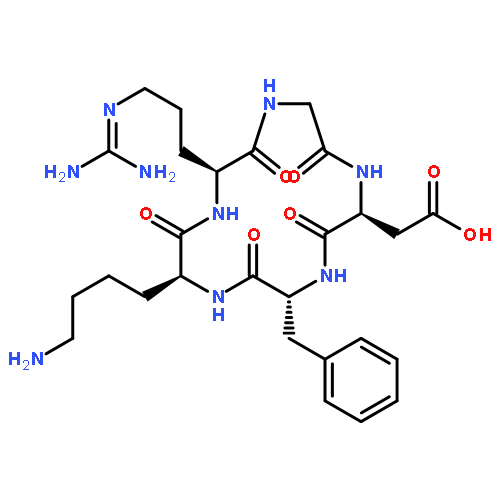
![2-[2-(2-METHOXYETHOXY)ETHOXYMETHYL]OXIRANE](http://img.cochemist.com/ccimg/71800/71712-93-1.png)
![2-[2-(2-METHOXYETHOXY)ETHOXYMETHYL]OXIRANE](http://img.cochemist.com/ccimg/71800/71712-93-1_b.png)


![POLY[OXY[(1R)-1-METHYL-2-OXO-1,2-ETHANEDIYL]]](http://img.cochemist.com/ccimg/27000/26917-25-9.png)
![POLY[OXY[(1R)-1-METHYL-2-OXO-1,2-ETHANEDIYL]]](http://img.cochemist.com/ccimg/27000/26917-25-9_b.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2_b.png)
![Poly[imino[(1S)-1-(carboxymethyl)-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26100/26063-13-8.png)
![Poly[imino[(1S)-1-(carboxymethyl)-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26100/26063-13-8_b.png)