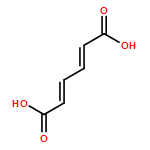
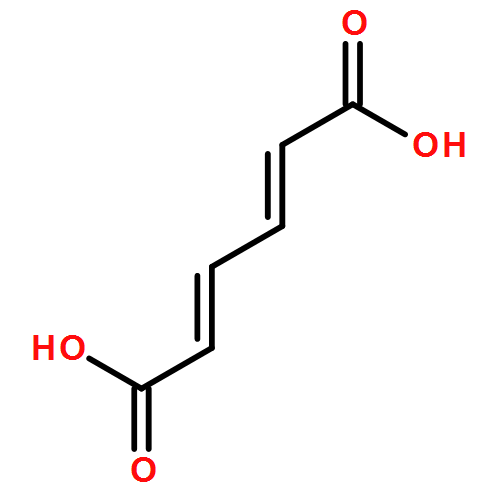
Co-reporter:Jixue Sun, Yang Li, Pi Liu, Jianping Lin
Journal of Molecular Graphics and Modelling 2017 Volume 74(Volume 74) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.jmgm.2017.04.009
•The bilayer could lead to a curvature induced by the transmembrane region of the Zika virus envelope protein.•The low pH condition could weaken the interactions and correlations in the envelope protein.•The highly conserved residues, His249, His288, His323 and His446 play key roles in driving the fusion process.The Zika virus has drawn worldwide attention because of the epidemic diseases it causes. It is a flavivirus that has an icosahedral protein shell constituted by an envelope glycoprotein (E-protein) and membrane protein (M-protein) in the mature virion. The multistep process of membrane fusion to infect the host cell is pH-induced. To understand the mechanism of the conformational changes in the (E-M)2 protein homodimer embedded in the membrane, two 200-ns accelerated dynamic simulations were performed under different pH conditions. The low pH condition weakens the interactions and correlations in both E-protein monomers and in the E-M heterodimer. The highly conserved residues, His249, His288, His323 and His446, are protonated under low pH conditions and play key roles in driving the fusion process. The analysis and discussion in this study may provide some insight into the molecular mechanism of Zika virus infection.Download high-res image (215KB)Download full-size image
Co-reporter:Jixue Sun, Yang Li, Jianping Lin
Journal of Molecular Graphics and Modelling 2017 Volume 74(Volume 74) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.jmgm.2017.03.003
•The single-stranded DNA could be easily adsorbed on the graphene.•DNA polymer could be adsorbed on the graphene by the flexible single-stranded DNA.•The adsorption could reduce the shape deformation of planar DNA polymers.DNA nanostructures can undergo large structural fluctuations and deviate from their intended configurations. In this work, two model DNA nanostructures (i.e., Nan and Kai) were designed based on the shape of the two Chinese characters of the name of Nankai University, and additional single-stranded DNA fragments were added to interact with graphene. During four 50-ns molecular dynamic simulations in aqueous solution, the DNA nanostructures adsorbed onto graphene demonstrated more stable conformations with lower root mean square deviations and smaller coordinate changes in the z-axis direction than the DNA nanostructures that were not adsorbed onto graphene. The interaction analyses and energetic calculations show that π-π interactions between single-stranded DNA and graphene are necessary for adsorption of the DNA nanostructures. Overall, this work examined the interactions between DNA and graphene at a large spatial scale with the hope that it provides a new strategy to stabilize DNA nanostructures.Download high-res image (168KB)Download full-size image
Co-reporter:Yang Li, Jixue Sun, Dongmei Li and Jianping Lin
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 18) pp:12642-12650
Publication Date(Web):04 Apr 2016
DOI:10.1039/C6CP00798H
The human glucagon receptor (GCGR) is a class B G-protein-coupled receptor (GPCR). The GCGR can be activated by glucagon and regulates the release of glucose. The GCGR has been proposed to be an important drug target for type 2 diabetes. Based on the structural model of a full-length glucagon-bound GCGR (glu-GCGR), we performed accelerated molecular dynamics (aMD) simulations, potential of mean force (PMF) calculations, cross-correlation analysis and community network analysis to study the activation mechanism and the conformational dynamics during the activation process. The PMF map depicts three different conformational states of the GCGR: the inactive, intermediate and active states. The activation of the GCGR is characterized by the outward movement of the intracellular side of helix VI. In the active state of the GCGR, the Arg1732.46–Ser3506.41 and Glu2453.50–Thr3516.42 hydrogen bonds break, and the χ1 rotamer of Phe3225.54 changes from perpendicular to parallel to helix VI. The binding of the agonist glucagon decreases the correlated motions of the extracellular loops (ELCs) and the helices around the glucagon-binding site. During the activation of the GCGR, the connections between the intracellular sides of helices become weaker, and the connections between glucagon and ECLs and the extracellular sides of helices become stronger. These facilitate G-protein coupling on the intracellular side and glucagon binding on the extracellular side, and stabilize the GCGR in the active state. We expect that this study can provide useful information on the activation mechanism of the GCGR and facilitate the future design of GCGR inhibitors.
Co-reporter:Yu Wei, Jinlong Li, Zeming Chen, Fengwei Wang, Weiqiang Huang, Zhangyong Hong, Jianping Lin
European Journal of Medicinal Chemistry 2015 Volume 101() pp:409-418
Publication Date(Web):28 August 2015
DOI:10.1016/j.ejmech.2015.06.054
•We apply an optimized hierarchical multistage virtual screening method.•The multistage VS method can have better performance than single VS methods.•The multistage VS approach dramatically increases the VS efficiency and accuracy.•We obtain two HIV-1 protease hits at micromolar range.The HIV-1 protease has proven to be a crucial component of the HIV replication machinery and a reliable target for anti-HIV drug discovery. In this study, we applied an optimized hierarchical multistage virtual screening method targeting HIV-1 protease. The method sequentially applied SVM (Support Vector Machine), shape similarity, pharmacophore modeling and molecular docking. Using a validation set (270 positives, 155,996 negatives), the multistage virtual screening method showed a high hit rate and high enrichment factor of 80.47% and 465.75, respectively. Furthermore, this approach was applied to screen the National Cancer Institute database (NCI), which contains 260,000 molecules. From the final hit list, 6 molecules were selected for further testing in an in vitro HIV-1 protease inhibitory assay, and 2 molecules (NSC111887 and NSC121217) showed inhibitory potency against HIV-1 protease, with IC50 values of 62 μM and 162 μM, respectively. With further chemical development, these 2 molecules could potentially serve as HIV-1 protease inhibitors.By using an optimized hierarchical multistage VS method, we obtained 2 compounds showing inhibition against HIV-1 protease at the μM level.
Co-reporter:Junli Xu, Zhonghua Wang, Pi Liu, Dongmei Li and Jianping Lin
Molecular BioSystems 2015 vol. 11(Issue 7) pp:2042-2050
Publication Date(Web):01 May 2015
DOI:10.1039/C5MB00159E
The human corticotropin-releasing factor receptor type 1 (CRF1R) is a class B G-protein-coupled receptor (GPCR), which mediates the response to stress and has been considered as a drug target for depression and anxiety. Based on the CRF1R-antagonist crystal structure, we study the binding mechanism of two distinct antagonists, CP-376395 and MTIP, and the dynamics behaviors of CRF1R induced by an antagonist binding. Key residues interacting with both antagonists and residues specifically binding to one of them are identified. Both antagonists interact with Asn283, Phe203, Met206, Leu280, Tyr316, Leu323, Leu287, Phe284, Val279, Leu319, Phe207, Gly210 and Phe362. CP-376395 specifically binds to Glu209 and Phe160, while MTIP specifically binds to Leu320, Leu213, Ile290, Phe162 and Val313. The total binding free energy of MTIP is lower than that of CP-376395; this is consistent with the experimental observation that MTIP shows higher binding affinity than CP-376395. The conformational dynamic behaviors of antagonist bound holo-CRF1R were found to be different from those of apo-CRF1R in three aspects: (i) the “ionic lock” between side chains of Arg151 in TM2 and Glu209 in TM3 was broken in apo-CRF1R, but was formed in holo-CRF1Rs; (ii) Phe203 in TM3 and Tyr327 in TM6 were in close proximity to each other in apo-CRF1R, while they were far apart resulting from the shift of TM6 in holo-CRF1Rs; and (iii) the “rotamer toggle switch”, Tyr327/Leu323/Phe284, adopted different rotameric conformations in apo-CRF1R and holo-CRF1Rs. We hope that our results could be helpful in further development of the drug design of CRF1R.
Co-reporter:Dongmei Li, Cui Liu, Jianping Lin
Journal of Molecular Graphics and Modelling 2015 Volume 55() pp:25-32
Publication Date(Web):February 2015
DOI:10.1016/j.jmgm.2014.10.014
•The mechanism of PAD4 inhibition by F-amidine was investigated using the QM/MM approach.•State I can transform to State N through a stepwise manner.•In the PAD4-F–amidine reactant complex, the active site Cys645 exists as a thiolate and His471 is protonated (i.e. State I).•The inhibition reaction proceeds through a three-membered sulfonium ring transition state, His471 acts as a proton donor and facilitates the inhibition reaction.Protein arginine deiminase 4 (PAD4) catalyzes the hydrolysis of a peptidylarginine residue to form a citrulline residue and ammonia during posttranslational modification. This process plays a pivotal role in rheumatoid arthritis (RA) and gene regulation. F-amidine belongs to a series of haloacetamidine compounds that are the most potent PAD4 inhibitors described to date. F-amidine acts as a mechanism-based inhibitor of PAD4, inactivating PAD4 by the covalent modification of the active site Cys645. In this manuscript, the fundamental mechanism of PAD4 inhibition by F-amidine is investigated using a QM/MM approach. Our simulations show that in the PAD4–F-amidine reactant complex, the active site Cys645 exists as a thiolate and His471 is protonated. This is consistent with the reverse protonation mechanism wherein the active site nucleophile, Cys645, in PAD4 exists as a thiolate in the active form of the enzyme. Inhibition of PAD4 by F-amidine is initiated by the nucleophilic addition of Sγ to the Cζ of F-amidine, leading to the formation of a tetrahedral intermediate. His471 serves as a proton donor, helping F to leave the fluoroacetamidine moiety of F-amidine; meanwhile, Sγ forms a three-membered ring with Cζ and Cη of F-amidine. Subsequently, the three-membered sulfonium ring collapses and rearranges to the final thioether product. His471 acts as a proton donor in the transition state and facilitates the inhibition reaction of PAD4.In the PAD4–F-amidine reactant complex, the active site Cys645 exists as a thiolate and His471 is protonated. The inhibition reaction proceeds through a three-membered sulfonium ring transition state, His471 acts as a proton donor and facilitates the inhibition.
Co-reporter:Zhihui Yan, Lijie Zhang, Haiyang Fu, Zhonghua Wang, Jianping Lin
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 2) pp:539-547
Publication Date(Web):15 January 2014
DOI:10.1016/j.bmcl.2013.12.026
With the emergence of drug resistance and the structural determination of the PA N-terminal domain (PAN), influenza endonucleases have become an attractive target for antiviral therapies for influenza infection. Here, we combined 3D-QSAR with side-chain hopping and molecular docking to produce novel structures as endonuclease inhibitors. First, a new molecular library was generated with side-chain hopping on an existing template molecule, L-742001, using an in-house fragment library that targets bivalent-cation-binding proteins. Then, the best 3D-QSAR model (AAAHR.500), with q2 = 0.76 and r2 = 0.97 from phase modeling, was constructed from 23 endonuclease inhibitors and validated with 17 test compounds. The AAAHR.500 model was then used to select effective candidates from the new molecular library. Combining 3D-QSAR with docking using Glide and Autodock, 13 compounds were considered the most likely candidate inhibitors. Docking studies showed that the binding modes of these compounds were consistent with the crystal structures of known inhibitors. These compounds could serve as potential endonuclease inhibitors for further biological activity tests.
Co-reporter:Feng Sang, Peng Feng, Jie Chen, Yahui Ding, Xiyan Duan, Jiadai Zhai, Xiaoyan Ma, Bin Zhang, Quan Zhang, Jianping Lin, Yue Chen
European Journal of Medicinal Chemistry 2013 Volume 68() pp:321-332
Publication Date(Web):October 2013
DOI:10.1016/j.ejmech.2013.08.003
•A highly efficient approach was applied to synthesize Epo D and its new analogues.•The new analogues demonstrated significant difference in biological activity.•The conformations of Epo D and its new analogues were investigated.Epothilone D (Epo D) and its 9-Methyl conformational analogues were synthesized through a highly efficient combinatorial approach. The fragment E was synthesized in 11 total steps with 6 longest linear steps, and each aldehyde B was prepared via a 3-step sequence. Starting from the common precursor E and a suitable aldehydes B, each target molecule were obtained in only 4 steps. The 9-(S)-epo D and 9-(R)-epo D demonstrated significant difference in inhibition activities against cancer cell lines and in conformational analysis.A highly efficient combinatorial approach was applied to synthesize epothilone D and its 9-Methyl analogues.
Co-reporter:Tong Huo;Yinjie Zhang
Protein & Cell 2012 Volume 3( Issue 8) pp:602-608
Publication Date(Web):2012 August
DOI:10.1007/s13238-012-2914-8
The giant panda is one of the most critically endangered species due to the fragmentation and loss of its habitat. Studying the functions of proteins in this animal, especially specific trait-related proteins, is therefore necessary to protect the species. In this work, the functions of these proteins were investigated using the genome sequence of the giant panda. Data on 21,001 proteins and their functions were stored in the Giant Panda Protein Database, in which the proteins were divided into two groups: 20,179 proteins whose functions can be predicted by GeneScan formed the known-function group, whereas 822 proteins whose functions cannot be predicted by GeneScan comprised the unknown-function group. For the known-function group, we further classified the proteins by molecular function, biological process, cellular component, and tissue specificity. For the unknown-function group, we developed a strategy in which the proteins were filtered by cross-Blast to identify panda-specific proteins under the assumption that proteins related to the panda-specific traits in the unknown-function group exist. After this filtering procedure, we identified 32 proteins (2 of which are membrane proteins) specific to the giant panda genome as compared against the dog and horse genomes. Based on their amino acid sequences, these 32 proteins were further analyzed by functional classification using SVM-Prot, motif prediction using MyHits, and interacting protein prediction using the Database of Interacting Proteins. Nineteen proteins were predicted to be zinc-binding proteins, thus affecting the activities of nucleic acids. The 32 panda-specific proteins will be further investigated by structural and functional analysis.
Co-reporter:Yu Wei, Ruihua Liu, Cui Liu, Jin Jin, Dongmei Li, Jianping Lin
Journal of Molecular Graphics and Modelling (March 2017) Volume 72() pp:88-95
Publication Date(Web):March 2017
DOI:10.1016/j.jmgm.2016.11.016
Co-reporter:Yang Li, Jixue Sun, Dongmei Li and Jianping Lin
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 18) pp:NaN12650-12650
Publication Date(Web):2016/04/04
DOI:10.1039/C6CP00798H
The human glucagon receptor (GCGR) is a class B G-protein-coupled receptor (GPCR). The GCGR can be activated by glucagon and regulates the release of glucose. The GCGR has been proposed to be an important drug target for type 2 diabetes. Based on the structural model of a full-length glucagon-bound GCGR (glu-GCGR), we performed accelerated molecular dynamics (aMD) simulations, potential of mean force (PMF) calculations, cross-correlation analysis and community network analysis to study the activation mechanism and the conformational dynamics during the activation process. The PMF map depicts three different conformational states of the GCGR: the inactive, intermediate and active states. The activation of the GCGR is characterized by the outward movement of the intracellular side of helix VI. In the active state of the GCGR, the Arg1732.46–Ser3506.41 and Glu2453.50–Thr3516.42 hydrogen bonds break, and the χ1 rotamer of Phe3225.54 changes from perpendicular to parallel to helix VI. The binding of the agonist glucagon decreases the correlated motions of the extracellular loops (ELCs) and the helices around the glucagon-binding site. During the activation of the GCGR, the connections between the intracellular sides of helices become weaker, and the connections between glucagon and ECLs and the extracellular sides of helices become stronger. These facilitate G-protein coupling on the intracellular side and glucagon binding on the extracellular side, and stabilize the GCGR in the active state. We expect that this study can provide useful information on the activation mechanism of the GCGR and facilitate the future design of GCGR inhibitors.

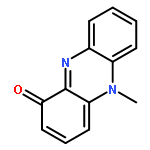
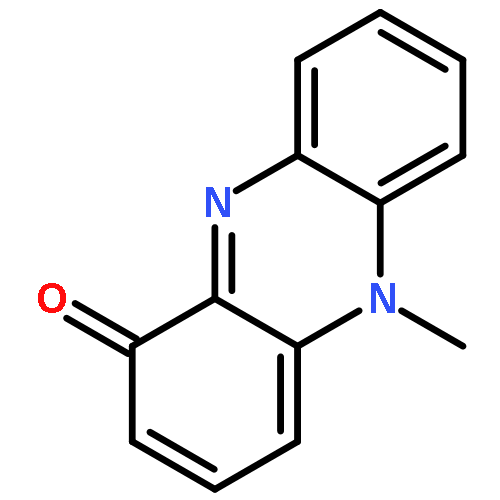








![L-Leucine,N-[(1,1-dimethylethoxy)carbonyl]-L-phenylalanyl-, methyl ester](http://img.cochemist.com/ccimg/64200/64152-76-7.png)
![L-Leucine,N-[(1,1-dimethylethoxy)carbonyl]-L-phenylalanyl-, methyl ester](http://img.cochemist.com/ccimg/64200/64152-76-7_b.png)
![Glycine, N-[1-[(1,1-dimethylethoxy)carbonyl]-L-prolyl]-, ethyl ester](http://img.cochemist.com/ccimg/57300/57294-31-2.png)
![Glycine, N-[1-[(1,1-dimethylethoxy)carbonyl]-L-prolyl]-, ethyl ester](http://img.cochemist.com/ccimg/57300/57294-31-2_b.png)
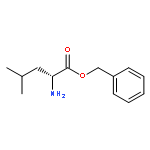
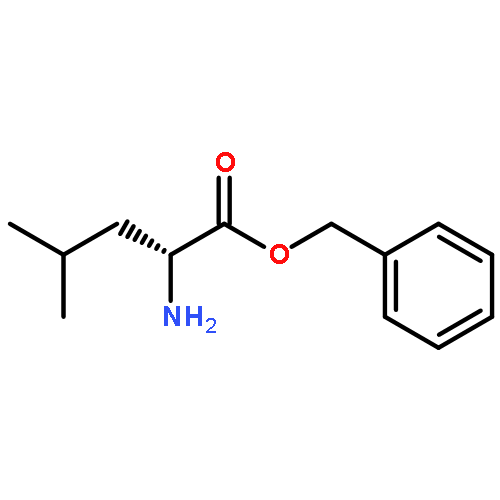
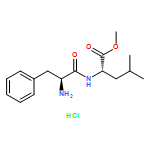
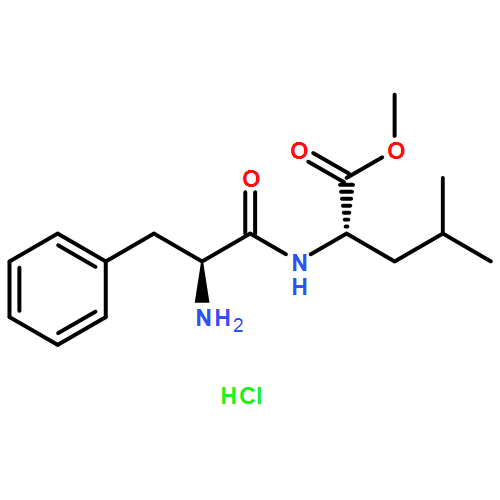
![methyl N-[(benzyloxy)carbonyl]serylleucinate](http://img.cochemist.com/ccimg/17400/17331-94-1.png)
![methyl N-[(benzyloxy)carbonyl]serylleucinate](http://img.cochemist.com/ccimg/17400/17331-94-1_b.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-phenylalanyl-, methyl ester](/data/chemimg/1085000/13122-89-9.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-phenylalanyl-, methyl ester](/data/chemimg/1085000/13122-89-9_b.png)
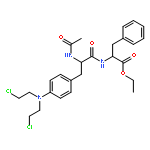
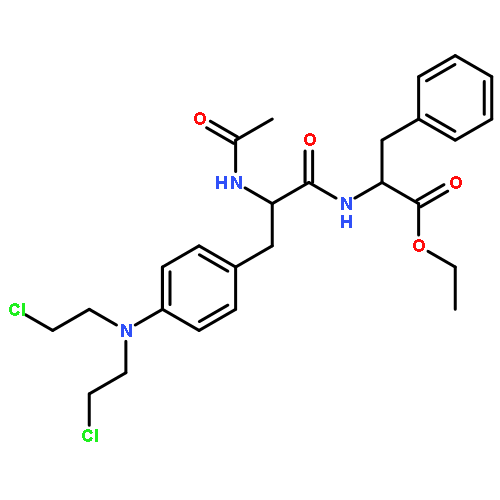
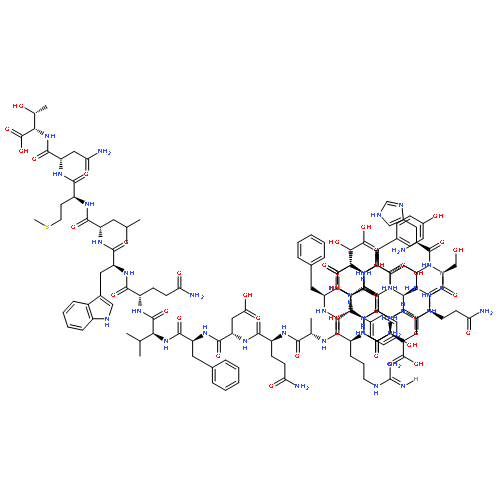
![L-Leucine, N-[(phenylmethoxy)carbonyl]-L-seryl-](http://img.cochemist.com/ccimg/2800/2768-55-0.png)
![L-Leucine, N-[(phenylmethoxy)carbonyl]-L-seryl-](http://img.cochemist.com/ccimg/2800/2768-55-0_b.png)