Co-reporter:Benoit Carbain ; David J. Paterson ; Elizabeth Anscombe ; Allyson J. Campbell ; Celine Cano ; Aude Echalier ; Jane A. Endicott ; Bernard T. Golding ; Karen Haggerty ; Ian R. Hardcastle ; Philip J. Jewsbury ; David R. Newell ; Martin E. M. Noble ; Celine Roche ; Lan Z. Wang ;Roger J. Griffin
Journal of Medicinal Chemistry 2014 Volume 57(Issue 1) pp:56-70
Publication Date(Web):December 4, 2013
DOI:10.1021/jm401555v
Evaluation of the effects of purine C-8 substitution within a series of CDK1/2-selective O6-cyclohexylmethylguanine derivatives revealed that potency decreases initially with increasing size of the alkyl substituent. Structural analysis showed that C-8 substitution is poorly tolerated, and to avoid unacceptable steric interactions, these compounds adopt novel binding modes. Thus, 2-amino-6-cyclohexylmethoxy-8-isopropyl-9H-purine adopts a “reverse” binding mode where the purine backbone has flipped 180°. This provided a novel lead chemotype from which we have designed more potent CDK2 inhibitors using, in the first instance, quantum mechanical energy calculations. Introduction of an ortho-tolyl or ortho-chlorophenyl group at the purine C-8 position restored the potency of these “reverse” binding mode inhibitors to that of the parent 2-amino-6-cyclohexylmethoxy-9H-purine. By contrast, the corresponding 8-(2-methyl-3-sulfamoylphenyl)-purine derivative exhibited submicromolar CDK2-inhibitory activity by virtue of engineered additional interactions with Asp86 and Lys89 in the reversed binding mode, as confirmed by X-ray crystallography.
Co-reporter:M. Raymond V. Finlay, Roger J. Griffin
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 17) pp:5352-5359
Publication Date(Web):1 September 2012
DOI:10.1016/j.bmcl.2012.06.053
Modulation of DNA repair pathways in oncology has been an area of intense interest in the last decade, not least as a consequence of the promising clinical activity of poly(ADP-ribose) polymerase (PARP) inhibitors. In this review article, we highlight inhibitors of the phosphatidylinositol 3-kinase related kinase (PIKK) family as of potential interest in the treatment of cancer, both in combination with DNA-damaging therapies and as stand-alone agents.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Kappusamy Saravanan ; Hannah C. Barlow ; Marion Barton ; A. Hilary Calvert ; Bernard T. Golding ; David R. Newell ; Julian S. Northen ; Nicola J. Curtin ; Huw D. Thomas ;Roger J. Griffin
Journal of Medicinal Chemistry 2011 Volume 54(Issue 6) pp:1847-1859
Publication Date(Web):March 2, 2011
DOI:10.1021/jm101493z
Membrane transport of nucleosides or nucleobases is mediated by transporters including the equilibrative nucleoside transporters (ENTs), and resistance to antitumor antimetabolite drugs may arise via salvage of exogenous purine or pyrimidine nucleosides or nucleobases by ENT transporters. The therapeutic utility of dipyridamole (3), a potent ENT inhibitor, is compromised by binding to the serum protein α1-acid glycoprotein (AGP). Derivatives and prodrugs of the ENT inhibitor 4,8-bis[(3,4-dimethoxybenzyl)amino]-2,6-bis[(2-hydroxypropyl)amino]pyrimido[5,4-d]pyrimidine (6, NU3108) are described, with improved in vivo pharmacokinetic properties and reduced AGP binding relative to dipyridamole. The mono- and diglycine carbamate derivatives were at least as potent as 6 and showed no reduction in potency by AGP. In a [3H]thymidine incorporation assay, employing COR-L23 cells, the diastereoisomers of 6 (IC50 = 26 nM) exhibited activity comparable with 3 (IC50 = 15 nM). The monophenyl carbamate and mono-4-methoxyphenyl carbamate exhibited the best ENT-inhibitory activity in the COR-L23 assay (IC50 = 8 and 4 nM, respectively). All of the new prodrugs were also highly effective at reversing thymidine/hypoxanthine rescue from pemetrexed cytotoxicity in the COR-L23 cell line.
Co-reporter:Sara L. Payne, Sonsoles Rodriguez-Aristegui, Julia Bardos, Céline Cano, Bernard T. Golding, Ian R. Hardcastle, Marcus Peacock, Nahida Parveen, Roger J. Griffin
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 12) pp:3649-3653
Publication Date(Web):15 June 2010
DOI:10.1016/j.bmcl.2010.04.102
Replacement of the core heterocycle of a defined series of chromen-4-one DNA-PK inhibitors by the isomeric chromen-2-one (coumarin) and isochromen-1-one (isocoumarin) scaffolds was investigated. Structure–activity relationships for DNA-PK inhibition were broadly consistent, albeit with a reduction of potency compared with the parent chromenone.Replacement of the core heterocycle of a defined series of chromen-4-one DNA-PK inhibitors by the isomeric chromen-2-one (coumarin) and isochromen-1-one (isocoumarin) scaffolds was investigated. Structure-activity relationships for DNA-PK inhibition were broadly consistent, albeit with a reduction of potency compared with the parent chromenone.
Co-reporter:Marine Desage-El Murr, Celine Cano, Bernard T. Golding, Ian R. Hardcastle, Marc Hummersome, Mark Frigerio, Nicola J. Curtin, Keith Menear, Caroline Richardson, Graeme C.M. Smith, Roger J. Griffin
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 17) pp:4885-4890
Publication Date(Web):1 September 2008
DOI:10.1016/j.bmcl.2008.07.066
The synthesis and biological evaluation of libraries of 8-biarylchromen-4-ones enabled the elucidation of structure–activity relationships for inhibition of the DNA-dependent protein kinase (DNA-PK), with 8-(3-(thiophen-2-yl)phenyl)chromen-4-one and 8-(3-(thiophen-3-yl)phenyl)chromen-4-one being especially potent inhibitors.The synthesis and biological evaluation of libraries of 8-biarylchromen-4-ones enabled the elucidation of structure–activity relationships for inhibition of the DNA-dependent protein kinase (DNA-PK), with 8-(3-(thiophen-2-yl)phenyl)chromen-4-one and 8-(3-(thiophen-3-yl)phenyl)chromen-4-one being especially potent inhibitors.
Co-reporter:Alex W. White, Nicola J. Curtin, Brian W. Eastman, Bernard T. Golding, Zdenek Hostomsky, Suzanne Kyle, Jianke Li, Karen A. Maegley, Donald J. Skalitzky, Stephen E. Webber, Xiao-Hong Yu, Roger J. Griffin
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 10) pp:2433-2437
Publication Date(Web):17 May 2004
DOI:10.1016/j.bmcl.2004.03.017
The synthesis and biological evaluation of a new series of amine-substituted 2-arylbenzimidazole-4-carboxamide inhibitors of the DNA-repair enzyme poly(ADP-ribose) polymerase-1 (PARP-1) is reported. The introduction of an amine substituent at the 2-aryl position is not detrimental to activity, with most inhibitors exhibiting Ki values for PARP-1 inhibition in the low nanomolar range. Two compounds in this series were found to potentiate the cytotoxicity of the DNA-methylating agent temozolomide by 4–5-fold in a human colorectal cancer cell line.The synthesis and biological evaluation of a new series of amine-substituted 2-arylbenzimidazole-4-carboxamide inhibitors of the DNA-repair enzyme poly(ADP-ribose) polymerase-1 (PARP-1) is reported. The introduction of an amine substituent at the 2-aryl position is not detrimental to activity, with most inhibitors exhibiting Ki values for PARP-1 inhibition in the low nanomolar range. Two compounds in this series were found to potentiate the cytotoxicity of the DNA-methylating agent temozolomide by 4–5-fold in a human colorectal cancer cell line.
Co-reporter:Justin J.J. Leahy, Bernard T. Golding, Roger J. Griffin, Ian R. Hardcastle, Caroline Richardson, Laurent Rigoreau, Graeme C.M. Smith
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 24) pp:6083-6087
Publication Date(Web):20 December 2004
DOI:10.1016/j.bmcl.2004.09.060
A solution-phase multiple-parallel synthesis approach was employed for the preparation of 6-, 7- and 8-aryl-substituted chromenone libraries, which were screened as inhibitors of the DNA repair enzyme DNA-dependent protein kinase (DNA-PK). These studies resulted in the identification of 8-dibenzothiophen-4-yl-2-morpholin-4-yl-chromen-4-one (NU7441) as a highly potent and selective DNA-PK inhibitor (IC50 = 14 nM), exhibiting ATP-competitive inhibition kinetics.A solution-phase multiple-parallel synthesis approach was employed for the preparation of 6-, 7- and 8-aryl-substituted chromenone libraries, which were screened as inhibitors of the DNA repair enzyme DNA-dependent protein kinase (DNA-PK). These studies resulted in the identification of 8-dibenzothiophen-4-yl-2-morpholin-4-yl-chromen-4-one (NU7441) as a highly potent and selective DNA-PK inhibitor (IC50 = 14 nM), exhibiting ATP-competitive inhibition kinetics.
Co-reporter:Kerry L. Sayle, Johanne Bentley, F.Thomas Boyle, A.Hilary Calvert, Yuzhu Cheng, Nicola J. Curtin, Jane A. Endicott, Bernard T. Golding, Ian R. Hardcastle, Philip Jewsbury, Veronique Mesguiche, David R. Newell, Martin E.M. Noble, Rachel J. Parsons, David J. Pratt, Lan Z. Wang, Roger J. Griffin
Bioorganic & Medicinal Chemistry Letters 2003 Volume 13(Issue 18) pp:3079-3082
Publication Date(Web):15 September 2003
DOI:10.1016/S0960-894X(03)00651-6
A series of O4-cyclohexylmethyl-5-nitroso-6-aminopyrimidines bearing 2-arylamino substituents was synthesised and evaluated for CDK1 and CDK2 inhibitory activity. Consistent with analogous studies with O6-cyclohexylmethylpurines, 2-arylaminopyrimidines with a sulfonamide or carboxamide group at the 4′-position were potent inhibitors, with IC50 values against CDK2 of 1.1±0.3 and 34±8 nM, respectively. The crystal structure of the 4′-carboxamide derivative, in complex with phospho-Thr160 CDK2/cyclin A, confirmed the expected binding mode of the inhibitor, and revealed an additional interaction between the carboxamide function and an aspartate residue.A series of O4-cyclohexylmethyl-5-nitroso-6-aminopyrimidines bearing 2-arylamino substituents was synthesised and evaluated for CDK1 and CDK2 inhibitory activity. Consistent with analogous studies with O6-cyclohexylmethylpurines, 2-arylaminopyrimidines with a sulfonamide or carboxamide group at the 4′-position were potent inhibitors, with IC50 values against CDK2 of 1.1±0.3 and 35±8 nM, respectively. The crystal structure of the 4′-carboxamide derivative, in complex with phospho-Thr160 CDK2/cyclin A, confirmed the expected binding mode of the inhibitor, and revealed an additional interaction between the carboxamide function and an aspartate residue.
Co-reporter:Jonathan J. Hollick, Bernard T. Golding, Ian R. Hardcastle, Niall Martin, Caroline Richardson, Laurent J.M. Rigoreau, Graeme C.M. Smith, Roger J. Griffin
Bioorganic & Medicinal Chemistry Letters 2003 Volume 13(Issue 18) pp:3083-3086
Publication Date(Web):15 September 2003
DOI:10.1016/S0960-894X(03)00652-8
6-Aryl-2-morpholin-4-yl-4H-pyran-4-ones and 6-aryl-2-morpholin-4-yl-4H-thiopyran-4-ones were synthesised and evaluated as potential inhibitors of the DNA repair enzyme DNA-dependent protein kinase (DNA-PK). Several compounds in each series exhibited superior activity to the chromenone LY294002, and were of comparable potency to the benzochromenone NU7026 (IC50=0.23 μM). Importantly, members of both structural classes were found to be selective inhibitors of DNA-PK over related phosphatidylinositol 3-kinase-related kinase (PIKK) family members. A multiple-parallel synthesis approach, employing Suzuki cross-coupling methodology, was utilised to prepare libraries of thiopyran-4-ones with a range of aromatic groups at the 3′- and 4′-positions on the thiopyran-4-one 6-aryl ring. Screening of the libraries resulted in the identification of 6-aryl-2-morpholin-4-yl-4H-thiopyran-4-ones bearing naphthyl or benzo[b]thienyl substituents at the 4′-position, as potent DNA-PK inhibitors with IC50 values in the 0.2–0.4 μM range.6-Aryl-2-morpholin-4-yl-4H-pyran-4-ones and 6-aryl-2-morpholin-4-yl-4H-thiopyran-4-ones were synthesised and evaluated as potential inhibitors of the DNA repair enzyme DNA-dependent protein kinase (DNA-PK). Several compounds in each series exhibited superior activity to the chromenone LY294002, and were of comparable potency to the benzochromenone NU7026 (IC50=0.23 μM). Importantly, members of both structural classes were found to be selective inhibitors of DNA-PK over related phosphatidylinositol 3-kinase-related kinase (PIKK) family members. A multiple-parallel synthesis approach, employing Suzuki cross-coupling methodology, was utilized to prepare libraries of thiopyran-4-ones with a range of aromatic groups at the 3′- and 4′-positions on the thiopyran-4-one 6-aryl ring. Screening of the libraries resulted in the identification of 6-aryl-2-morpholin-4-yl-4H-thiopyran-4-ones bearing naphthyl or benzo[b]thienyl substituents at the 4′-position, as potent DNA-PK inhibitors with IC50 values in the 0.2–0.4 μM range.
Co-reporter:Martin Stockley, William Clegg, Gabriele Fontana, Bernard T Golding, Niall Martin, Laurent J.M Rigoreau, Graeme C.M Smith, Roger J Griffin
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 21) pp:2837-2841
Publication Date(Web):5 November 2001
DOI:10.1016/S0960-894X(01)00537-6
The first reported synthesis of the DNA-PK inhibitor 3-cyano-6-hydrazonomethyl-5-(4-pyridyl)pyrid-[1H]-2-one (OK-1035) is described. The structure of OK-1035 was validated by X-ray crystallography. An IC50 value of 100 μM was determined for inhibition of DNA-PK, and this is approximately 12-fold higher than that reported previously.The first reported synthesis of the DNA-PK inhibitor 3-cyano-6-hydrazonomethyl-5-(4-pyridyl)pyrid-[1H]-2-one (OK-1035) is described. The structure of OK-1035 was validated by X-ray crystallography. An IC50 value of 100 μM was determined for inhibition of DNA-PK, and this is approximately 12-fold higher than that reported previously.
Co-reporter:Hannah C Barlow, Karen J Bowman, Nicola J Curtin, A.Hilary Calvert, Bernard T Golding, Bing Huang, Peter J Loughlin, David R Newell, Peter G Smith, Roger J Griffin
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 6) pp:585-589
Publication Date(Web):20 March 2000
DOI:10.1016/S0960-894X(00)00053-6
The synthesis and biological evaluation of potent 4,8-dibenzylaminopyrimidopyrimidine nucleoside transport inhibitors, with reduced binding to α1-acid glycoprotein, is reported.





![Glycine, N-[[(3-cyano-4,6-dimethyl-2-pyridinyl)thio]acetyl]-](http://img.cochemist.com/ccimg/385500/385417-58-3.png)
![Glycine, N-[[(3-cyano-4,6-dimethyl-2-pyridinyl)thio]acetyl]-](http://img.cochemist.com/ccimg/385500/385417-58-3_b.png)
![Glycine, N-[[[3-cyano-4-(4-fluorophenyl)-6-phenyl-2-pyridinyl]thio]acetyl]-](http://img.cochemist.com/ccimg/353500/353463-50-0.png)
![Glycine, N-[[[3-cyano-4-(4-fluorophenyl)-6-phenyl-2-pyridinyl]thio]acetyl]-](http://img.cochemist.com/ccimg/353500/353463-50-0_b.png)





















![2-Propen-1-one, 1-phenyl-3-[4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/144100/144017-74-3.png)
![2-Propen-1-one, 1-phenyl-3-[4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/144100/144017-74-3_b.png)


























![4-[2-(4-METHYL-1-PIPERAZINYL)ETHOXY]ANILINE](http://img.cochemist.com/ccimg/39000/38948-28-6.png)
![4-[2-(4-METHYL-1-PIPERAZINYL)ETHOXY]ANILINE](http://img.cochemist.com/ccimg/39000/38948-28-6_b.png)















![2-Amino-N,N-diethylbenzo[d]thiazole-6-sulfonamide](http://img.cochemist.com/ccimg/18000/17901-14-3.png)
![2-Amino-N,N-diethylbenzo[d]thiazole-6-sulfonamide](http://img.cochemist.com/ccimg/18000/17901-14-3_b.png)







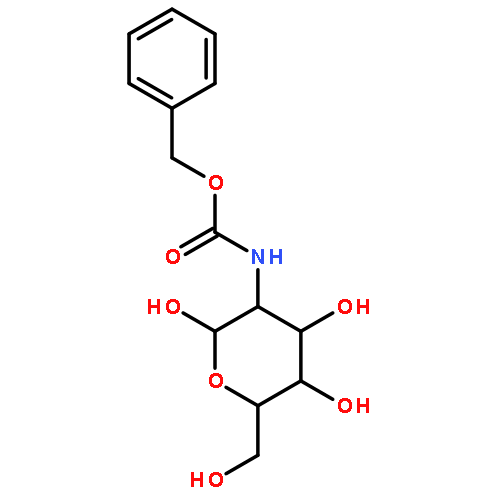






![Methyl 2-{[(benzyloxy)carbonyl]amino}-2-deoxy-α-d-glucopyranoside](http://img.cochemist.com/ccimg/4800/4704-15-8.png)
![Methyl 2-{[(benzyloxy)carbonyl]amino}-2-deoxy-α-d-glucopyranoside](http://img.cochemist.com/ccimg/4800/4704-15-8_b.png)