Co-reporter:Maureen E. Smith, Sara L. Johnson, and Douglas S. Masterson
Journal of Chemical Education January 8, 2013 Volume 90(Issue 1) pp:
Publication Date(Web):November 2, 2012
DOI:10.1021/ed300053z
A two-part demonstration was conducted in our first-semester organic chemistry course designed to introduce students to the formation of alcohols, regioselective reactions, and analysis of organic products by NMR analysis. This demonstration utilized the oxymercuration–demercuration sequence to prepare an alcohol from an alkene in a Markovnikov manner because the reaction is easy to execute and has a dramatic, observable color change during the transformation. The alcohol product produced was then utilized in a classroom demonstration of 1H NMR using a remote accessible NMR spectrometer.Keywords: Constitutional Isomers; Demonstrations; Hands-On Learning/Manipulatives; NMR Spectroscopy; Organic Chemistry; Second-Year Undergraduate;
Co-reporter:Hari Kiran Kotapati;Jamarii D. Robinson;Daniel R. Lawrence;Kimberly R. Fortner;Caleb W. Stanford;Douglas R. Powell;Rainer Wardenga;Uwe T. Bornscheuer;Douglas S. Masterson
European Journal of Organic Chemistry 2017 Volume 2017(Issue 20) pp:3009-3016
Publication Date(Web):2017/05/26
DOI:10.1002/ejoc.201700605
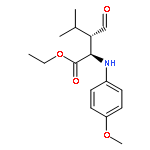
Malonate diesters with highly branched side chains containing a preexisting chiral center were prepared from optically pure amino alcohols and subjected to asymmetric enzymatic hydrolysis by Porcine Liver Esterase (PLE). Recombinant PLE isoenzymes have been utilized in this work to synthesize diastereomerically enriched malonate half-esters from enantiopure malonate diesters. The diastereomeric excess of the product half-esters was further improved in the later steps of synthesis either by simple recrystallization or flash column chromatography. The diastereomerically enriched half-ester was transformed into a novel 5-substituted Cα-methyl-β-proline analogue (3R,5S)-1c, in high optical purity employing a stereoselective cyclization methodology. This β-proline analogue was tested for activity as a catalyst of the Mannich reaction. The β-proline analogue derived from the hydrolysis reaction by the crude PLE appeared to catalyze the Mannich reaction between an α-imino ester and an aldehyde providing decent to good diastereoselectivities. However, the enantioselectivities in the reaction was low. The second diastereomer of the 5-benzyl-substituted Cα-methyl-β-proline, (3S,5S)-1c was prepared by enzymatic hydrolysis using PLE isoenzyme 3 and tested for its catalytic activity in the Mannich reaction. Amino acid, (3S,5S)-1c catalyzed the Mannich reaction between isovaleraldehyde and an α-imino ester yielding the “anti” selective product with an optical purity of 99 %ee.
Co-reporter:Hari Kiran Kotapati, Daniel R. Lawrence, Shelby O. Thames, Douglas S. Masterson
Tetrahedron Letters 2016 Volume 57(Issue 39) pp:4389-4391
Publication Date(Web):28 September 2016
DOI:10.1016/j.tetlet.2016.08.052
•Enantioselective synthesis of both enantiomers of NH-Fmoc-S-Trityl-Cα-Methyl Cysteine in >99%ee.•Convenient protocols for the protecting group installations.•Curtius rearrangement to prepare Fmoc protected amine using Ti(IV)Isopropoxide.•Mercury(II) mediated quick S-tert-butyl deprotection.•Recrystallization to provide optically pure NH-Fmoc-S-Trityl-Cα-Methyl Cysteine.A concise, chiral auxiliary free, methodology for the preparation of orthogonally protected Cα-Methyl Cysteine has been developed. Curtius rearrangement of malonate halfester from PLE hydrolysis (J. Pept. Sci.2008, 14, 1151; J. Org. Chem.2003, 68, 5403) and simultaneous Fmoc protection using Titanium(IV) Isopropoxide yielded Fmoc protected amino ester that is then transformed into (R)-NH-Fmoc-S-Trityl-Cα-Methyl Cysteine in three steps. For the synthesis of the S enantiomer of the protected amino acid, enantiomerically enriched CO2-t-Bu half ester derived from PLE hydrolysis was subjected to Curtius reaction followed by Fmoc protection using Titanium(IV) Isopropoxide which is then converted to (S)-NH-Fmoc-S-Trityl-Cα-Methyl Cysteine in two steps.Figure options
Co-reporter:Souvik Banerjee, Emily R. Vogel, Daniel Hinton, Michael Sterling, Douglas S. Masterson
Tetrahedron: Asymmetry 2015 Volume 26(21–22) pp:1292-1299
Publication Date(Web):1 December 2015
DOI:10.1016/j.tetasy.2015.09.014
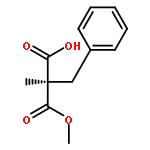
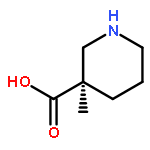
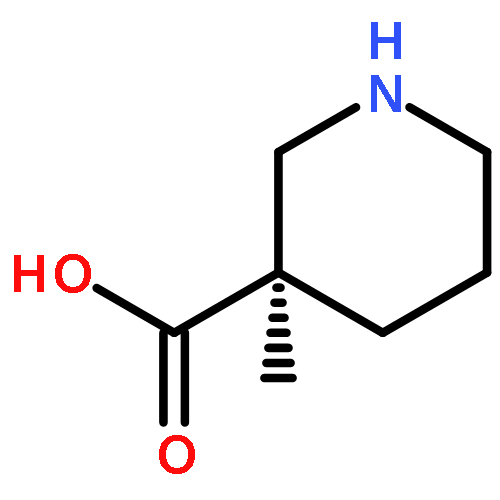
A stereoselective and enantiodivergent strategy for the construction of δ-lactams is described. The strategy utilizes chiral malonic esters prepared from enantiomerically enriched mono esters of disubstituted malonic acid. A cyclization occurs with the selective displacement of a substituted benzyl alcohol as the leaving group. The resulting δ-lactams are then converted into nipecotic acid analogues using straightforward transformations. The resulting nipecotic acid analogues proved capable organocatalysts in Mannich reactions.(R)-5-(1,3-Dioxoisoindolin-2-yl)-2-(ethoxycarbonyl)-2-methylpentanoic acidC17H19NO6[α]D24 = +5.8 (c 2, MeOH)Source of chirality: EnzymaticAbsolute configuration: (R)(S)-1-Ethyl 3-(4-nitrobenzyl) 2-(3-(1,3-dioxoisoindolin-2-yl)propyl)-2-methylmalonateC24H24N2O8[α]D23 = −33 (c 1, CH2Cl2)Source of chirality: the precursorAbsolute configuration: (S)(R)-Ethyl 3-methyl-2-oxopiperidine-3-carboxylateC9H15NO3[α]D24 = +29.2 (c 1, CH2Cl2)Source of chirality: the precursorAbsolute configuration: (R)(R)-Ethyl 1-benzyl-3-methyl-2-oxopiperidine-3-carboxylateC16H21NO3[α]D24 = +62.6 (c 2, CH2Cl2)Source of chirality: the precursorAbsolute configuration: (R)(S)-Ethyl 1-benzyl-3-methyl-2-thioxopiperidine-3-carboxylateC16H21NO2S[α]D22 = +75.2 (c 1, CH2Cl2)Source of chirality: the precursorAbsolute configuration: (S)(R)-Ethyl 1-benzyl-3-methylpiperidine-3-carboxylateC16H23NO2[α]D21 = +11.8 (c 1, CH2Cl2)Source of chirality: the precursorAbsolute configuration: (R)(R)-1-Benzyl-3-methylpiperidine-3-carboxylic acidC14H19NO2[α]D22 = +19.0 (c 1, MeOH)Source of chirality: the precursorAbsolute configuration: (R)(R)-3-Methylpiperidine-3-carboxylic acidC7H13NO2[α]D24 = +1.2 (c 1, MeOH)Source of chirality: the precursorAbsolute configuration: (R)(S)-3-Methylpiperidine-3-carboxylic acidC7H13NO2[α]D21 = −1.5 (c 1, MeOH)Source of chirality: the precursorAbsolute configuration: (S)(S)-1-tert-Butyl 3-ethyl 2-(3-(1,3-dioxoisoindolin-2-yl)propyl)-2-methylmalonateC21H27NO6[α]D23 = −5.2 (c 1, MeOH)Source of chirality: the precursorAbsolute configuration: (S)(S)-tert-Butyl 3-methyl-2-oxopiperidine-3-carboxylateC11H19NO3[α]D23 = −16.2 (c 1, CH2Cl2)Source of chirality: the precursorAbsolute configuration: (S)
Co-reporter:Maureen E. Smith;Steven A. Knolls
Journal of The American Society for Mass Spectrometry 2015 Volume 26( Issue 3) pp:397-403
Publication Date(Web):2015 March
DOI:10.1007/s13361-014-1041-6
A mass spectrometry assay is presented here that allows for the simultaneous determination of yield and enantioselectivity in a single analysis. The assay makes use of molecules that are structurally similar to the analytes of interest as standards. The assay predicts the yields of the reactions reasonably well and with little error. For example, in the pig liver esterase catalyzed hydrolysis of one prochiral malonate, the yield predicted by the assay was 72%, while larger scale isolated reaction yields were within 5% of this value. This assay provides a fast method to determine yield and enantioselectivity in one analysis. The strengths and limitations of this method are discussed.
Co-reporter:Souvik Banerjee, Walker J. Wiggins, Jessie L. Geoghegan, Catherine T. Anthony, Eugene A. Woltering and Douglas S. Masterson
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 37) pp:6307-6319
Publication Date(Web):05 Aug 2013
DOI:10.1039/C3OB41282B
Prochiral malonic diesters containing a quaternary carbon center have been successfully transformed into a diverse set of tBoc-Fmoc-α2,2-methyllysine-OH analogues through chiral malonic half-ester intermediates obtained via enzymatic (Pig Liver Esterase, PLE) hydrolysis. The variety of chiral half-ester intermediates, which vary from 1 to 6 methylene units in the side chain, are achieved in moderate to high optical purity and in good yields. The PLE hydrolysis of malonic diesters with various side chain lengths appears to obey the Jones's PLE model according to the stereochemical configurations of the resulting chiral half-esters. The established synthetic strategy allows the construction of both enantiomers of α2,2-methyllysine analogues, and a (S)-β2,2-methyllysine analogue from a common synthon by straightforward manipulation of protecting groups. Two different straightforward and cost effective synthetic strategies are described for the synthesis of α2,2-methyllysine analogues. The described strategies should find significant usefulness in preparing novel peptide libraries with unnatural lysine analogues. A Vapreotide analogue incorporating (S)-α2,2-methyllysine was prepared. However, the Vapreotide analogue with (S)-α-methyl-α-lysine is found to lose its specific binding to somatostatin receptor subtype 2 (SSTR2).
Co-reporter:Souvik Banerjee, Justin Smith, Jillian Smith, Caleb Faulkner, and Douglas S. Masterson
The Journal of Organic Chemistry 2012 Volume 77(Issue 23) pp:10925-10930
Publication Date(Web):November 5, 2012
DOI:10.1021/jo3015903
A straightforward stereoselective and enantiodivergent cyclization strategy for the construction of γ-lactams is described. The cyclization strategy makes use of chiral malonic esters prepared from enantiomerically enriched monoesters of disubstituted malonic acid. The cyclization occurs with the selective displacement of a substituted benzyl alcohol as the leaving group. A Hammett study illustrates that the cyclization is under electronic control. The resulting γ-lactam can be readily converted into a novel proline analogue.
Co-reporter:Maureen E. Smith;Souvik Banerjee;Yongliang Shi;Dr. Marlen Schmidt; Uwe T. Bornscheuer; Douglas S. Masterson
ChemCatChem 2012 Volume 4( Issue 4) pp:472-475
Publication Date(Web):
DOI:10.1002/cctc.201100490
Co-reporter:Dale A. Rosado Jr., Tina S. Masterson, and Douglas S. Masterson
Journal of Chemical Education 2011 Volume 88(Issue 2) pp:178-183
Publication Date(Web):November 12, 2010
DOI:10.1021/ed100043m
Mass spectrometry (MS) has been gaining in popularity in recent years owing in large part to the development of soft-ionization techniques such as matrix-assisted laser desorption ionization (MALDI) and electrospray ionization (ESI). These soft-ionization techniques have opened up the field of MS analysis to biomolecules, polymers, and other high molecular weight materials. The growing publication rate and a quick survey of available job postings requiring MS skill sets illustrate the importance of properly training students in the use of modern MS techniques. Many colleges and universities are unable to hire full-time technical staff devoted to student training on the use of MS equipment. The lack of technical staff results in less than adequate student training on the use of MS equipment, which ultimately leads to student apprehension about using the instruments regularly in their research projects. Herein, we report on our efforts utilizing a mini-session course format (one week with 40 contact hours) to properly train graduate students in the use of modern MS equipment and techniques. The course consists of three components: (i) a lecture component of approximately 2 h each day to introduce the basic theory of the days topics, (ii) a hands-on laboratory component illustrating the use of both MALDI and ESI instruments to gather a variety of data, and (iii) a student project utilizing the MS equipment in their own graduate research during the regular semester following the mini-session course. The students receive three hours of graduate credit and receive permission, upon successful completion of the course, to use the MS facilities unsupervised. The faculty member teaching the course receives teaching credit for the training efforts.Keywords (Audience): Continuing Education; Graduate Education/Research; Keywords (Domain): Curriculum; Laboratory Instruction; Keywords (Pedagogy): Hands-On Learning/Manipulatives; Keywords (Topic): Applications of Chemistry; Bioanalytical Chemistry; Bioorganic Chemistry; Mass Spectrometry;
Co-reporter:Douglas S. Masterson, Dale A. Rosado Jr., Cassie Nabors
Tetrahedron: Asymmetry 2009 Volume 20(Issue 13) pp:1476-1486
Publication Date(Web):16 July 2009
DOI:10.1016/j.tetasy.2009.05.037
A practical mass spectrometry-based enantioselectivity assay is presented which makes use of enantiomerically enriched, but not enantiomerically pure, probe molecules readily obtained from esterase hydrolysis of prochiral malonates. The technique presented here allows us to recycle materials obtained from esterase hydrolysis which give substantial, but synthetically insufficient, enantiomeric excess as probe molecules in an enantioselectivity assay. The enantiomerically enriched products are esterified using deuterium-labelled alcohol. The enantiomeric excess is measured using mass spectrometry (LC–MS and LDI) by measuring the D5/H5 ratio in the resulting products obtained from an enzymatic hydrolysis. The D5/H5 ratio is corrected to account for the enantiomeric purity of the probe. Herein we report the results obtained from Pig Liver Esterase hydrolyses of prochiral malonate esters and outline the strengths and limitations of this approach to enantioselectivity determinations. This assay strategy was able to identify reaction conditions that led to an improvement in ee from 70% ee to >97% ee in the PLE-catalyzed hydrolysis of a prochiral malonate used to prepare unnatural serine analogues.(R)-2-(4-(Benzyloxymethyl)-3-ethoxy-2-methyl-3-oxopropanoic acidC14H18O5[α]D21.8=+7.7 (c 0.208, MeOH)Source of chirality: asymmetric synthesisAbsolute configuration: (2R)(R)-2-(4-(Benzyloxy)benzyl)-3-ethoxy-2-methyl-3-oxopropanoic acidC20H22O5[α]D22=-1.0 (c 0.066, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2R)(S)-Ethyl-3-(benzyloxy)-2-((4-methoxybenzyloxy)carbonylamino)-2-methylpropanoateC28H31NO6[α]D17.8=+24.2 (c 0.07, CH2Cl2).Source of chirality: asymmetric synthesisAbsolute configuration: (1S)(S)-α-Methyl tyrosine ethyl esterC12H17NO3[α]obs21.7=-0.10 (c 1.2 M HCl)Source of chirality: asymmetric synthesisAbsolute configuration: (1S)
Co-reporter:Huiyong Yin;Almary Chacon;Ned A. Porter
Journal of The American Society for Mass Spectrometry 2007 Volume 18( Issue 5) pp:807-816
Publication Date(Web):2007 May
DOI:10.1016/j.jasms.2007.01.004
Protein identification is routinely accomplished by peptide sequencing using mass spectrometry (MS) after enzymatic digestion. Site-specific chemical modification may improve peptide ionization efficiency or sequence coverage in mass spectrometry. We report herein that amino group of lysine residue in peptides can be selectively modified by reaction with a peroxycarbonate and the resulting lysine peroxycarbamates undergo homolytic fragmentation under conditions of low-energy collision-induced dissociation (CID) in electrospray ionization (ESI) and matrix-assisted laser desorption and ionization (MALDI) MS. Selective modification of lysine residue in peptides by our strategy can induce specific peptide cleavage at or near the lysine site. Studies using deuterated analogues of modified lysine indicate that fragmentation of the modified peptides involves apparent free-radical processes that lead to peptide chain fragmentation and side-chain loss. The formation of a-, c-, or z-types of ions in MS is reminiscent of the proposed free-radical mechanisms in low-energy electron capture dissociation (ECD) processes that may have better sequence coverage than that of the conventional CID method. This site-specific cleavage of peptides by free radical- promoted processes is feasible and such strategies may aid the protein sequencing analysis and have potential applications in top-down proteomics.
Co-reporter:Huiyong Yin, Almary Chacon, Ned A. Porter, Douglas S. Masterson
Journal of the American Society for Mass Spectrometry (May 2007) Volume 18(Issue 5) pp:807-816
Publication Date(Web):1 May 2007
DOI:10.1016/j.jasms.2007.01.004
Protein identification is routinely accomplished by peptide sequencing using mass spectrometry (MS) after enzymatic digestion. Site-specific chemical modification may improve peptide ionization efficiency or sequence coverage in mass spectrometry. We report herein that amino group of lysine residue in peptides can be selectively modified by reaction with a peroxycarbonate and the resulting lysine peroxycarbamates undergo homolytic fragmentation under conditions of low-energy collision-induced dissociation (CID) in electrospray ionization (ESI) and matrix-assisted laser desorption and ionization (MALDI) MS. Selective modification of lysine residue in peptides by our strategy can induce specific peptide cleavage at or near the lysine site. Studies using deuterated analogues of modified lysine indicate that fragmentation of the modified peptides involves apparent free-radical processes that lead to peptide chain fragmentation and side-chain loss. The formation of a-, c-, or z-types of ions in MS is reminiscent of the proposed free-radical mechanisms in low-energy electron capture dissociation (ECD) processes that may have better sequence coverage than that of the conventional CID method. This site-specific cleavage of peptides by free radical– promoted processes is feasible and such strategies may aid the protein sequencing analysis and have potential applications in top-down proteomics.
Co-reporter:Souvik Banerjee, Walker J. Wiggins, Jessie L. Geoghegan, Catherine T. Anthony, Eugene A. Woltering and Douglas S. Masterson
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 37) pp:NaN6319-6319
Publication Date(Web):2013/08/05
DOI:10.1039/C3OB41282B
Prochiral malonic diesters containing a quaternary carbon center have been successfully transformed into a diverse set of tBoc-Fmoc-α2,2-methyllysine-OH analogues through chiral malonic half-ester intermediates obtained via enzymatic (Pig Liver Esterase, PLE) hydrolysis. The variety of chiral half-ester intermediates, which vary from 1 to 6 methylene units in the side chain, are achieved in moderate to high optical purity and in good yields. The PLE hydrolysis of malonic diesters with various side chain lengths appears to obey the Jones's PLE model according to the stereochemical configurations of the resulting chiral half-esters. The established synthetic strategy allows the construction of both enantiomers of α2,2-methyllysine analogues, and a (S)-β2,2-methyllysine analogue from a common synthon by straightforward manipulation of protecting groups. Two different straightforward and cost effective synthetic strategies are described for the synthesis of α2,2-methyllysine analogues. The described strategies should find significant usefulness in preparing novel peptide libraries with unnatural lysine analogues. A Vapreotide analogue incorporating (S)-α2,2-methyllysine was prepared. However, the Vapreotide analogue with (S)-α-methyl-α-lysine is found to lose its specific binding to somatostatin receptor subtype 2 (SSTR2).

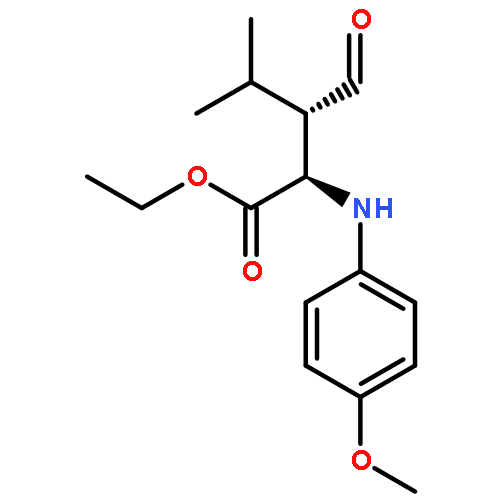
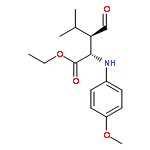

![Acetic acid, [(4-methoxyphenyl)imino]-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/120500/120419-96-7.png)
![Acetic acid, [(4-methoxyphenyl)imino]-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/120500/120419-96-7_b.png)