Co-reporter:S S Tarighat, R Santhanam, D Frankhouser, H S Radomska, H Lai, M Anghelina, H Wang, X Huang, L Alinari, A Walker, M A Caligiuri, C M Croce, L Li, R Garzon, C Li, R A Baiocchi and G Marcucci
Leukemia 2016 30(4) pp:789-799
Publication Date(Web):November 27, 2015
DOI:10.1038/leu.2015.308
Changes in the enzymatic activity of protein arginine methyltransferase (PRMT) 5 have been associated with cancer; however, the protein’s role in acute myeloid leukemia (AML) has not been fully evaluated. Here, we show that increased PRMT5 activity enhanced AML growth in vitro and in vivo while PRMT5 downregulation reduced it. In AML cells, PRMT5 interacted with Sp1 in a transcription repressor complex and silenced miR-29b preferentially via dimethylation of histone 4 arginine residue H4R3. As Sp1 is also a bona fide target of miR-29b, the miR silencing resulted in increased Sp1. This event in turn led to transcription activation of FLT3, a gene that encodes a receptor tyrosine kinase. Inhibition of PRMT5 via sh/siRNA or a first-in-class small-molecule inhibitor (HLCL-61) resulted in significantly increased expression of miR-29b and consequent suppression of Sp1 and FLT3 in AML cells. As a result, significant antileukemic activity was achieved. Collectively, our data support a novel leukemogenic mechanism in AML where PRMT5 mediates both silencing and transcription of genes that participate in a ‘yin-yang’ functional network supporting leukemia growth. As FLT3 is often mutated in AML and pharmacologic inhibition of PRMT5 appears feasible, the PRMT5–miR-29b–FLT3 network should be further explored as a novel therapeutic target for AML.
Co-reporter:Huameng Li ; Hui Xiao ; Li Lin ; David Jou ; Vandana Kumari ; Jiayuh Lin
Journal of Medicinal Chemistry 2014 Volume 57(Issue 3) pp:632-641
Publication Date(Web):January 23, 2014
DOI:10.1021/jm401144z
The IL-6/GP130/STAT3 pathway is critical for the progression of multiple types of cancers. We report here the discovery of raloxifene and bazedoxifene as novel inhibitors of IL-6/GP130 protein–protein interactions (PPIs) using multiple ligand simultaneous docking (MLSD) and drug repositioning approaches. Multiple drug scaffolds were simultaneously docked into hot spots of GP130 D1 domain by MLSD to compete with the key interacting residues of IL-6, followed by tethering to generate virtual hit compounds. Similarity searches of virtual hits on drug databases identified raloxifene and bazedoxifene as potential inhibitors of IL-6/GP130 interaction. In cancer cell assays both compounds bind to GP130 and demonstrated selective inhibition of IL-6 induced STAT3 phosphorylation and were significantly more potent than the previously reported natural product inhibitor MDL-A. The identified drugs represent a new class of lead compounds with piperidine, benzothiophene, and indole scaffolds to inhibit IL-6 induced homodimerization of GP130. Besides potential direct usage for clinic trials, the two compounds can also serve as lead compounds for optimization to speed the development of drugs selectively targeting the IL-6/GP130/STAT3 cancer signaling pathway.
Co-reporter:Wenying Yu ; Hui Xiao ; Jiayuh Lin
Journal of Medicinal Chemistry 2013 Volume 56(Issue 11) pp:4402-4412
Publication Date(Web):May 7, 2013
DOI:10.1021/jm400080c
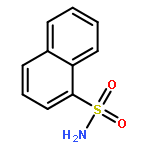
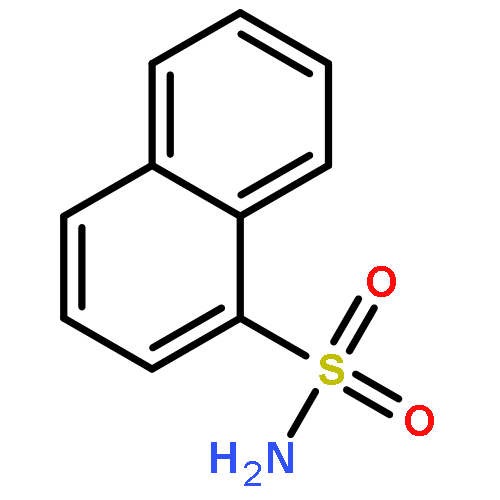
Constitutive activation of signal transducer and activator of transcription 3 (STAT3) has been validated as an attractive therapeutic target for cancer therapy. To stop both STAT3 activation and dimerization, a viable strategy is to design inhibitors blocking its SH2 domain phosphotyrosine binding site that is responsible for both actions. A new fragment-based drug design (FBDD) strategy, in silico site-directed FBDD, was applied in this study. A designed novel compound, 5,8-dioxo-6-(pyridin-3-ylamino)-5,8-dihydronaphthalene-1-sulfonamide (LY5), was confirmed to bind to STAT3 SH2 by fluorescence polarization assay. In addition, four out of the five chosen compounds have IC50 values lower than 5 μM for the U2OS cancer cells. 8 (LY5) has an IC50 range in 0.5–1.4 μM in various cancer cell lines. 8 also suppresses tumor growth in an in vivo mouse model. This study has demonstrated the utility of this approach and could be used to other drug targets in general.
Co-reporter:Somsundaram N. Chettiar, James V. Cooley, In-Hee Park, Deepak Bhasin, Arnab Chakravarti, Pui-Kai Li, Chenglong Li, Naduparambil Korah Jacob
Bioorganic & Medicinal Chemistry Letters 2013 23(19) pp: 5429-5433
Publication Date(Web):
DOI:10.1016/j.bmcl.2013.07.034
Co-reporter:Kiran V. Mahasenan and Chenglong Li
Journal of Chemical Information and Modeling 2012 Volume 52(Issue 5) pp:1345-1355
Publication Date(Web):April 29, 2012
DOI:10.1021/ci300040c
Kinase targets have been demonstrated to undergo major conformational reorganization upon ligand binding. Such protein conformational plasticity remains a significant challenge in structure-based virtual screening methodology and may be approximated by screening against an ensemble of diverse protein conformations. Maternal embryonic leucine zipper kinase (MELK), a member of serine-threonine kinase family, has been recently found to be involved in the tumerogenic state of glioblastoma, breast, ovarian, and colon cancers. We therefore modeled several conformers of MELK utilizing the available chemogenomic and crystallographic data of homologous kinases. We carried out docking pose prediction and virtual screening enrichment studies with these conformers. The performances of the ensembles were evaluated by their ability to reproduce known inhibitor bioactive conformations and to efficiently recover known active compounds early in the virtual screen when seeded with decoy sets. A few of the individual MELK conformers performed satisfactorily in reproducing the native protein–ligand pharmacophoric interactions up to 50% of the cases. By selecting an ensemble of a few representative conformational states, most of the known inhibitor binding poses could be rationalized. For example, a four conformer ensemble is able to recover 95% of the studied actives, especially with imperfect scoring function(s). The virtual screening enrichment varied considerably among different MELK conformers. Enrichment appears to improve by selection of a proper protein conformation. For example, several holo and unliganded active conformations are better to accommodate diverse chemotypes than ATP-bound conformer. These results prove that using an ensemble of diverse conformations could give a better performance. Applying this approach, we were able to screen a commercially available library of half a million compounds against three conformers to discover three novel inhibitors of MELK, one from each template. Among the three compounds validated via experimental enzyme inhibition assays, one is relatively potent (15; Kd = 0.37 μM), one moderately active (12; Kd = 3.2 μM), and one weak but very selective (9; Kd = 18 μM). These novel hits may be utilized to assist in the development of small molecule therapeutic agents useful in diseases caused by deregulated MELK, and perhaps more importantly, the approach demonstrates the advantages of choosing an appropriate ensemble of a few conformers in pursuing compound potency, selectivity, and novel chemotypes over using single target conformation for structure-based drug design in general.
Co-reporter:Huameng Li ; Aiguo Liu ; Zhenjiang Zhao ; Yufang Xu ; Jiayuh Lin ; David Jou
Journal of Medicinal Chemistry 2011 Volume 54(Issue 15) pp:5592-5596
Publication Date(Web):June 16, 2011
DOI:10.1021/jm101330h
We describe a novel method of drug discovery using MLSD and drug repositioning, with cancer target STAT3 being used as a test case. Multiple drug scaffolds were simultaneously docked into hot spots of STAT3 by MLSD, followed by tethering to generate virtual template compounds. Similarity search of virtual hits on drug database identified celecoxib as a novel inhibitor of STAT3. Furthermore, we designed two novel lead inhibitors based on one of the lead templates and celecoxib.
Co-reporter:Kiran V. Mahasenan, Ryan E. Pavlovicz, Brandon J. Henderson, Tatiana F. González-Cestari, Bitna Yi, Dennis B. McKay, and Chenglong Li
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 11) pp:855
Publication Date(Web):September 18, 2011
DOI:10.1021/ml2001714
We performed a hierarchical structure-based virtual screening utilizing a comparative model of the human α4β2 neuronal nicotinic acetylcholine receptor (nAChR) extracellular domain. Compounds were selected for experimental testing based on structural diversity, binding pocket location, and standard error of the free energy scoring function used in the screening. Four of the eleven in silico hit compounds showed promising activity with low micromolar IC50 values in a calcium accumulation assay. Two of the antagonists were also proven to be selective for human α4β2 vs human α3β4 nAChRs. This is the first report of successful discovery of novel nAChR antagonists through the use of structure-based virtual screening with a human nAChR homology model. These compounds may serve as potential novel scaffolds for further development of selective nAChR antagonists.Keywords: AChBP; allosteric; nAChR; nicotinic acetylcholine receptor; noncompetitive antagonists; virtual screening
Co-reporter:In-Hee Park
Journal of Molecular Recognition 2011 Volume 24( Issue 2) pp:254-265
Publication Date(Web):
DOI:10.1002/jmr.1047
Abstract
Signal transducer and activator of transcription 3 (STAT3) is an anti-cancer target protein due to its over-activation in tumor cells. The Tyr705-phosphorylated (pTyr) STAT3 binds to the pTyr-recognition site of its Src Homology 2 (SH2) domain of another STAT3 monomer to form a homo-dimer, which then causes cellular anti-apoptosis, proliferation, and tumor invasion. Recently, many STAT3 SH2 dimerization inhibitors have been discovered via both computational and experimental methods. To systematically assess their binding affinities and specificities, for eight representative inhibitors, we utilized molecular docking, molecular dynamics simulation, and ensuing energetic analysis to compare their binding characteristics. The inhibitors' binding free energies were calculated via MMPB(GB)SA, and the STAT3 SH2 binding “hot spots” were evaluated through binding energy decomposition and hydrogen bond (H-bond) distribution analysis. Several conclusions can be drawn: (1) the overall enthalpy–entropy compensation paradigm is preserved for the STAT3 SH2/ligand binding thermodynamics; (2) at one end of the binding spectrum, two compounds bind to SH2 due to their minimum entropic penalties that result from their relative rigidities and increased dynamics of SH2 upon their binding; at the other end of the binding spectrum, one compound shows a typical weak binder behavior due to its loose binding in the SH2's strongest enthalpy-contributing binding subsite; (3) hydrogen bonding seems a strong indicator to evaluate the SH2/ligand binding potency, which echoes a finding that CH/π non-classical H-bond is responsible for some pTyr peptides binding to their corresponding SH2 domains; (4) STAT3 SH2 domain possesses three binding “hot spots”: pTyr705-binding pocket with polar residues and contributing the largest binding enthalpy (two-thirds); Leu706 subsite which is the most dynamic and hardest to target; a hydrophobic side pocket which is unique to STAT3 and very targetable, which may offer unique opportunity to design STAT3-specific inhibitors, particularly with fragment-based approach. Copyright © 2010 John Wiley & Sons, Ltd.
Co-reporter:In-Hee Park and Chenglong Li
The Journal of Physical Chemistry B 2010 Volume 114(Issue 15) pp:5144-5153
Publication Date(Web):March 25, 2010
DOI:10.1021/jp911085d
Survivin is an anticancer drug target due to its overexpression in tumor cells in a homodimer form. Abbott Laboratories has identified a small molecule binding site near the dimerization interface in a high-throughput-screening (HTS)-NMR experiment. A benchmarking of the binding mode of the compound Abbott8 aided in the search for the ligand-induced-fit receptor structure by exploring the conformational space of the survivin dimer. We performed ensemble dockings with Abbott8 against a large set of conformations sampled via replica exchange molecular dynamics (REMD). This enhanced sampling allowed the reproduction of the holo-NMR experimental binding mode. Surprisingly, the major structural change in the best-REMD snapshot corresponding to the small molecule induced-fit happens in the so-called “survivin mitosis/apoptosis switch loop”, consistent with the X-ray crystal structure of survivin-monomer/borealin/INCENP chromosomal passenger complex (CPC), as the distance between Phe93 and Phe101 increased. To verify this hypothetical pathway for the induced-fit conformational change, we utilized morphed intermediate structures that combined the X-ray data and the best-REMD snapshot, and the potential of mean force (PMF) of the survivin dimer was constructed with umbrella sampling (US) followed by a multiple Bennett acceptance ratio estimator (MBAR). It revealed a 3−4 kcal/mol free energy barrier along the reaction coordinate, and the complex is stabilized by the gain of the binding energies of Abbott8. This free energy barrier might prohibit the reproduction of the experimental binding mode from the regular NTP-MD ensemble docking that we had tried. The combination of REMD generalized ensemble sampling with ensemble docking and free energy pathway analysis may provide a novel research protocol for the simulation of protein−ligand induced-fit recognition.

![1H-Indol-5-ol, 1-[[4-[2-(hexahydro-1H-azepin-1-yl)ethoxy]phenyl]methyl]-2-(4-hydroxyphenyl)-3-methyl-](/data/chemimg/128100/198481-32-2.png)
![1H-Indol-5-ol, 1-[[4-[2-(hexahydro-1H-azepin-1-yl)ethoxy]phenyl]methyl]-2-(4-hydroxyphenyl)-3-methyl-](/data/chemimg/128100/198481-32-2_b.png)