Co-reporter:Larissa Kuznetsova, Liang Sun, Jin Chen, Xianrui Zhao, Joshua Seitz, Manisha Das, Yuan Li, Jean M. Veith, Paula Pera, Ralph J. Bernacki, Shujun Xia, Susan B. Horwitz, Iwao Ojima
Journal of Fluorine Chemistry 2012 Volume 143() pp:177-188
Publication Date(Web):November 2012
DOI:10.1016/j.jfluchem.2012.07.007
A series of 3′-difluorovinyl taxoids with C10 modifications, as well as those with C2 and C10 modifications, were strategically designed to block the metabolism by cytochrome P-450 3A4 enzyme and synthesized. These novel difluorovinyl taxoids were evaluated for their cytotoxicity against drug-sensitive human breast (MCF7), multidrug-resistant (MDR) human ovarian (NCI/ADR), human colon (HT-29) and human pancreatic (PANC-1) cancer cell lines. 3′-Difluorovinyl taxoids exhibit several to 16 times better activity against MCF7, HT-29 and PANC-1 cell lines and up to three orders of magnitude higher potency against NCI/ADR cell line as compared to paclitaxel. Structure–activity relationship study shows the critical importance of the C2 modifications on the activity against MDR cancer cell line, while the C10 modifications have a rather minor effect on the potency with some exceptions. The effect of the C2 modifications on potency against MCF7 cell line increases in the following order: H < F < Cl < N3. Among the twenty five 3′-difluorovinyl taxoids evaluated, eight taxoids exhibited less than 100 pM IC50 values against MCF7 cell line. Difluorovinyl taxoids induced GTP-independent tubulin polymerization much faster than paclitaxel. Then, the resulting microtubules were stable to Ca2+-induced depolymerization, indicating strong stabilization of microtubules. Molecular modeling study indicated that a difluorovinyl taxoid binds to β-tubulin in a manner that is consistent with the REDOR-Taxol structure. The difluorovinyl group appears to mimic the isobutenyl group to some extent, but with very different electronic property, which may account for the unique activities of difluorovinyl taxoids.Graphical abstractNovel 3′-difluorovinyl taxoids were synthesized and evaluated for their in vitro cytotoxicities against human breast, ovarian, colon and pancreatic cancer cell lines. These taxoids exhibit three orders of magnitude higher potency than paclitaxel against multidrug-resistant cancer cell lines. Structure–activity relationship, tubulin polymerization/microtubule stabilization activity, and molecular modeling study of these highly potent taxoids are discussed.Highlights► A series of 3′-difluorovinyl taxoids were strategically designed to block the metabolism by P-450 3A4 enzyme and synthesized. ► Difluorovinyl taxoids exhibit up to 3 orders of magnitude higher potency against MDR cell line as compared to paclitaxel. ► Difluorovinyl taxoids induced GTP-independent tubulin polymerization much faster than paclitaxel. ► Molecular modeling study indicates that a difluorovinyl taxoid binds to β-tubulin consistent with the REDOR-Taxol structure. ► Difluorovinyl group's unique stereoelectronic property may account for the high potency of difluorovinyl taxoids.
Co-reporter:Anushree Kamath, Iwao Ojima
Tetrahedron 2012 68(52) pp: 10640-10664
Publication Date(Web):
DOI:10.1016/j.tet.2012.07.090
Co-reporter:Kunal Kumar ; Divya Awasthi ; Seung-Yub Lee ; Ilaria Zanardi ; Bela Ruzsicska ; Susan Knudson ; Peter J. Tonge ; Richard A. Slayden
Journal of Medicinal Chemistry 2011 Volume 54(Issue 1) pp:374-381
Publication Date(Web):December 2, 2010
DOI:10.1021/jm1012006
Libraries of novel trisubstituted benzimidazoles were created through rational drug design. A good number of these benzimidazoles exhibited promising MIC values in the range of 0.5−6 μg/mL (2−15 μM) for their antibacterial activity against Mtb H37Rv strain. Moreover, five of the lead compounds also exhibited excellent activity against clinical Mtb strains with different drug-resistance profiles. All lead compounds did not show appreciable cytotoxicity (IC50 > 200 μM) against Vero cells, which inhibited Mtb FtsZ assembly in a dose dependent manner. The two lead compounds unexpectedly showed enhancement of the GTPase activity of Mtb FtsZ. The result strongly suggests that the increased GTPase activity destabilizes FtsZ assembly, leading to efficient inhibition of FtsZ polymerization and filament formation. The TEM and SEM analyses of Mtb FtsZ and Mtb cells, respectively, treated with a lead compound strongly suggest that lead benzimidazoles have a novel mechanism of action on the inhibition of Mtb FtsZ assembly and Z-ring formation.
Co-reporter:Ce Shi;Chih-Wei Chien ;Dr. Iwao Ojima
Chemistry – An Asian Journal 2011 Volume 6( Issue 2) pp:674-680
Publication Date(Web):
DOI:10.1002/asia.201000697
Abstract
A library of new 2,2′-bis(diphenylphosphinoyloxy)-1,1′-binaphthyl (binapo)-type chiral diphosphonite ligands was designed and synthesized based on chiral 3,3′,5,5′,6,6′-hexasubstituted biphenols. These bop ligands have exhibited excellent efficiency in a palladium-catalyzed intermolecular allylic amination reaction, which provides a key intermediate for the total synthesis of Strychnos indole alkaloids with enantiopurities of up to 96 % ee.
Co-reporter:Chi-Feng Lin and Iwao Ojima
The Journal of Organic Chemistry 2011 Volume 76(Issue 15) pp:6240-6249
Publication Date(Web):June 14, 2011
DOI:10.1021/jo2009615
Formal enantioselective total synthesis of schulzeines A–C was accomplished, featuring highly efficient Pd-catalyzed asymmetric allylic amination using novel diphosphonite ligands (BOPs) to provide 1-vinyltetrahydroisoquinoline key intermediates, as well as Ru-catalyzed ring-closing metathesis reaction to construct the key tricyclic cores in enantiopure form with correct absolute configurations.
Co-reporter:Shuyi Chen, Xianrui Zhao, Jingyi Chen, Jin Chen, Larisa Kuznetsova, Stanislaus S. Wong and Iwao Ojima
Bioconjugate Chemistry 2010 Volume 21(Issue 5) pp:979
Publication Date(Web):April 29, 2010
DOI:10.1021/bc9005656
An efficient mechanism-based tumor-targeting drug delivery system, based on tumor-specific vitamin-receptor mediated endocytosis, has been developed. The tumor-targeting drug delivery system is a conjugate of a tumor-targeting molecule (biotin: vitamin H or vitamin B-7), a mechanism-based self-immolative linker and a second-generation taxoid (SB-T-1214) as the cytotoxic agent. This conjugate (1) is designed to be (i) specific to the vitamin receptors overexpressed on tumor cell surface and (ii) internalized efficiently through receptor-mediated endocytosis, followed by smooth drug release via glutathione-triggered self-immolation of the linker. In order to monitor and validate the sequence of events hypothesized, i.e., receptor-mediated endocytosis of the conjugate, drug release, and drug-binding to the target protein (microtubules), three fluorescent/fluorogenic molecular probes (2, 3, and 4) were designed and synthesized. The actual occurrence of these processes was unambiguously confirmed by means of confocal fluorescence microscopy (CFM) and flow cytometry using L1210FR leukemia cells, overexpressing biotin receptors. The molecular probe 4, bearing the taxoid linked to fluorescein, was also used to examine the cell specificity (i.e., efficacy of receptor-based cell targeting) for three cell lines, L1210FR (biotin receptors overexpressed), L1210 (biotin receptors not overexpressed), and WI38 (normal human lung fibroblast, biotin receptor negative). As anticipated, the molecular probe 4 exhibited high specificity only to L1210FR. To confirm the direct correlation between the cell-specific drug delivery and anticancer activity of the probe 4, its cytotoxicity against these three cell lines was also examined. The results clearly showed a good correlation between the two methods. In the same manner, excellent cell-specific cytotoxicity of the conjugate 1 (without fluorescein attachment to the taxoid) against the same three cell lines was confirmed. This mechanism-based tumor-targeting drug delivery system will find a range of applications.
Co-reporter:Liang Sun, Jean M. Veith, Paula Pera, Ralph J. Bernacki, Iwao Ojima
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 19) pp:7101-7112
Publication Date(Web):1 October 2010
DOI:10.1016/j.bmc.2010.07.069
Novel paclitaxel-mimicking alkaloids were designed and synthesized based on a bioactive conformation of paclitaxel, that is, REDOR-Taxol. The alkaloid 2 bearing a 5-7-6 tricyclic scaffold mimics REDOR-Taxol best among the compounds designed and was found to be the most potent compound against several drug-sensitive and drug-resistant human cancer cell lines. MD simulation study on the paclitaxel mimics 1 and 2 as well as REDOR-Taxol bound to the 1JFF tubulin structure was quite informative to evaluate the level of mimicking. The MD simulation study clearly distinguishes the 5-6-6 and 5-7-6 tricyclic scaffolds, and also shows substantial difference in the conformational stability of the tubulin-bound structures between 2 and REDOR-Taxol. The latter may account for the large difference in potency, and provides critical information for possible improvement in the future design of paclitaxel mimics.Design, syntheses and biological evaluations of de novo paclitaxel mimics are reported.
Co-reporter:Joseph J. Kaloko, Yu-Han Gary Teng and Iwao Ojima
Chemical Communications 2009 (Issue 30) pp:4569-4571
Publication Date(Web):29 Jun 2009
DOI:10.1039/B909781C
Rapid construction of 5-7-6-5 fused tetracyclic carbocycles and heterocycles from cyclohexene-diynes and CO has been achieved in one step through a Rh(I)-catalyzed [2 + 2 + 2 + 1] cycloaddition process.
Co-reporter:Iwao Ojima and Manisha Das
Journal of Natural Products 2009 Volume 72(Issue 3) pp:554-565
Publication Date(Web):February 24, 2009
DOI:10.1021/np8006556
Among the numerous chemotherapeutic drugs, paclitaxel and docetaxel are among the most widely used against various types of cancer. However, these drugs cause undesirable side effects as well as drug resistance. Therefore, it is essential to develop “taxane” anticancer agents with better pharmacological properties and improved activity especially against drug-resistant cancers. Several laboratories have performed extensive SAR studies on paclitaxel. Our SAR studies have led to the development of numerous highly potent novel second- and third-generation taxoids with systematic modifications at the C-2, C-10, and C-3′ positions. The third-generation taxoids showed virtually no difference in potency against drug-resistant and drug-sensitive cell lines. Some of the new generation taxoids also exhibited excellent cytotoxicity against pancreatic cell lines expressing multidrug-resistant genes. We have also designed taxoids with strategic fluorine incorporation to investigate their effects on the cytotoxicity and the blockage of known metabolic pathways. Furthermore, we have successfully employed computational biology analysis to design novel macrocyclic taxoids to mimic the bioactive conformation of paclitaxel. This account describes our work on the design, synthesis, and biological evaluation of these novel taxoids, which has led to the discovery of very promising candidates for further preclinical studies.
Co-reporter:Antonella Pepe, Liang Sun, Ilaria Zanardi, Xinyuan Wu, Cristiano Ferlini, Gabriele Fontana, Ezio Bombardelli, Iwao Ojima
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 12) pp:3300-3304
Publication Date(Web):15 June 2009
DOI:10.1016/j.bmcl.2009.04.070
Novel C-seco-taxoids were synthesized from 10-deacetylbaccatin III and their potencies evaluated against drug-sensitive and drug-resistant cancer cell lines. The drug-resistant cell lines include ovarian cancer cell lines resistant to cisplatin, topotecan, adriamycin and paclitaxel overexpressing class III β-tubulin, A2780TC1 and A2780TC3. The last two cell lines were selected through chronic exposure of A2780wt to paclitaxel and Pgp blocker cyclosporine. All novel C-seco-taxoids exhibited remarkable potency against A2780TC1 and A2780TC3 cell lines, and no cross resistance to cisplatin- and topotecan-resistant cell lines, A2780CIS and A2780TOP. Four of those C-seco-taxoids exhibit much higher activities than IDN5390 against paclitaxel-resistant cell lines, A2780ADR, A2780TC1 and A2780TC3. SB-CST-10202 possesses the best all-round high potencies across different drug-resistant cell lines. Molecular modeling studies, including molecular dynamics simulations, on the drug-protein complexes of class I and III β-tubulins were performed to identify possible cause of the remarkable potency of these C-seco-taxoids against paclitaxel-resistant cell lines overexpressing class III β-tubulin.The syntheses and biological evaluations of a series of novel C-seco-taxoids against paclitaxel-resistant human ovarian cancer cell lines overexpressing class III β-tubulin and other drug-resistant phenotypes are reported.
Co-reporter:Liang Sun Dr.;Carlos Simmerling Dr. Dr.
ChemMedChem 2009 Volume 4( Issue 5) pp:719-731
Publication Date(Web):
DOI:10.1002/cmdc.200900044
Co-reporter:Iwao Ojima
Accounts of Chemical Research 2008 Volume 41(Issue 1) pp:108
Publication Date(Web):July 31, 2007
DOI:10.1021/ar700093f
A long-standing problem in cancer chemotherapy is the lack of tumor-specific treatments. Traditional chemotherapy relies on the premise that rapidly proliferating cancer cells are more likely to be killed by a cytotoxic agent. In reality, however, cytotoxic agents have very little or no specificity, which leads to systemic toxicity, causing undesirable severe side effects. Therefore, the development of innovative and efficacious tumor-specific drug delivery protocols or systems is urgently needed. A rapidly growing tumor requires various nutrients and vitamins. Thus, tumor cells overexpress many tumor-specific receptors, which can be used as targets to deliver cytotoxic agents into tumors. This Account presents our research program on the discovery and development of novel and efficient drug delivery systems, possessing tumor-targeting ability and efficacy against various cancer types, especially multidrug-resistant tumors. In general, a tumor-targeting drug delivery system consists of a tumor recognition moiety and a cytotoxic warhead connected directly or through a suitable linker to form a conjugate. The conjugate, which can be regarded as a “guided molecular missile”, should be systemically nontoxic, that is, the linker must be stable in blood circulation, but upon internalization into the cancer cell, the conjugate should be readily cleaved to regenerate the active cytotoxic warhead. These novel “guided molecular missiles” are conjugates of the highly potent second-generation taxoid anticancer agents with tumor-targeting molecules through mechanism-based cleavable linkers. These conjugates are specifically delivered to tumors and internalized into tumor cells, and the potent taxoid anticancer agents are released from the linker into the cytoplasm. We have successfully used omega-3 polyunsaturated fatty acids, in particular DHA, and monoclonal antibodies (for EGFR) as tumor-targeting molecules for the conjugates, which exhibited remarkable efficacy against human tumor xenografts in animal models. We have developed self-immolative disulfide linkers wherein the glutathione-triggered cascade drug release takes place to generate the original anticancer agent. The use of disulfide linkers is attractive beacuse it takes into account the fact that the concentration of glutathione is much higher (>1000 times) in tumor cells than in blood plasma. In order to monitor and elucidate the mechanism of tumor-targeting, internalization, and drug release, several fluorescent and fluorogenic probes using biotin as the tumor-targeting module were developed and used. Then, the progressive occurrence of the designed receptor-mediated endocytosis, drug release, and drug binding to the target protein (microtubules) has been successfully observed and confirmed by means of confocal fluorescence microscopy. These “guided molecular missiles” provide bright prospects for the development of highly efficacious new generation drugs for cancer chemotherapy.
Co-reporter:Iwao Ojima ; Jin Chen ; Liang Sun ; Christopher P. Borella ; Tao Wang ; Michael L. Miller ; Songnian Lin ; Xudong Geng ; Larisa Kuznetsova ; Chuanxing Qu ; David Gallager ; Xianrui Zhao ; Ilaria Zanardi ; Shujun Xia ; Susan B. Horwitz ; Jon Mallen-St. Clair ; Jennifer L. Guerriero ; Dafna Bar-Sagi ; Jean M. Veith ; Paula Pera ;Ralph J. Bernacki
Journal of Medicinal Chemistry 2008 Volume 51(Issue 11) pp:3203-3221
Publication Date(Web):May 9, 2008
DOI:10.1021/jm800086e
Novel second-generation taxoids with systematic modifications at the C2, C10, and C3′N positions were synthesized and their structure−activity relationships studied. A number of these taxoids exhibited exceptionally high potency against multidrug-resistant cell lines, and several taxoids exhibited virtually no difference in potency against the drug-sensitive and drug-resistant cell lines. These exceptionally potent taxoids were termed “third-generation taxoids”. 19 (SB-T-1214), 14g (SB-T-121303), and 14i (SB-T-1213031) exhibited excellent activity against paclitaxel-resistant ovarian cancer cell lines with mutations in β-tubulin as well, wherein the drug resistance is mediated by the β-tubulin mutation. These taxoids were found to possess exceptional activity in promoting tubulin assembly, forming numerous very short microtubules similar to those formed by discodermolide. Taxoids 19 and 14g also showed excellent cytotoxicity against four pancreatic cancer cell lines, expressing three to four multidrug-resistant genes. Moreover, taxoid 19 exhibited excellent in vivo efficacy against highly drug-resistant CFPAC-1 pancreatic as well as DLD-1 human colon tumor xenografts in mice.
Co-reporter:Larissa V. Kuznetsova, Antonella Pepe, Ioana M. Ungureanu, Paula Pera, Ralph J. Bernacki, Iwao Ojima
Journal of Fluorine Chemistry 2008 Volume 129(Issue 9) pp:817-828
Publication Date(Web):September 2008
DOI:10.1016/j.jfluchem.2008.05.013
A series of novel 3′-difluoromethyl-taxoids and 3′-trifluoromethyl-taxoids with modifications at the C2 and C10 positions were synthesized and evaluated for their in vitro cytotoxicities against human breast carcinoma (MCF7-S, MCF7-R, LCC6-WT, LCC6-MDR), non-small cell lung carcinoma (H460) and colon adenocarcinoma (HT-29) cell lines. These second-generation fluoro-taxoids exhibited several times to more than 20 times better potency than paclitaxel against drug-sensitive cancer cell lines, MCF7-S, LCC6-WT, H460, and HT-29. These fluoro-taxoids also possess two orders of magnitude higher potency than paclitaxel against drug-resistant cancer cell lines, MCF7-R and LCC6-MDR. Structure–activity relationship study shows the importance of the C10 modification for increasing the activity against multidrug-resistant cancer cell lines. Effects of the C2-benzoate modifications on the potency in the 3′-difluoromethyl-taxoid series are very clear (i.e., F < MeO < Cl < N3), while those in the 3′-trifluoromethyl-taxoid series are less obvious. Also, different trends in the sensitivity to the C2-substitution are observed between drug-sensitive cell lines and drug-resistant cancer cell lines that overexpress efflux pumps.Novel 3′-difluoromethyl-taxoids and 3′-trifluoromethyl-taxoids were synthesized and evaluated for their in vitro cytotoxicities against human breast, non-small cell lung, and colon cancer cell lines. These second-generation fluoro-taxoids exhibit two orders of magnitude higher potency than paclitaxel against multidrug-resistant cancer cell lines. Structure–activity relationship of these highly potent fluoro-taxoids is discussed.
Co-reporter:Larissa Kuznetsova, Jin Chen, Liang Sun, Xinyuan Wu, Antonella Pepe, Jean M. Veith, Paula Pera, Ralph J. Bernacki, Iwao Ojima
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 4) pp:974-977
Publication Date(Web):15 February 2006
DOI:10.1016/j.bmcl.2005.10.089
Polyunsaturated fatty acids such as docosahexaenoic acid (DHA), linolenic acid, and linoleic acid were linked to the C-2′ position of the second-generation taxoids that could overcome MDR caused by overexpressed ABC transporters. The new conjugates, tested in vivo, exhibited strong activity against drug-resistant colon cancer and drug-sensitive ovarian cancer xenografts in mice. Two of the new conjugates, DHA–SB-T-1214 and DHA–SB-T-1213, were found to achieve the total regression of drug-resistant and drug-sensitive tumors, respectively, in the animal models with substantially reduced systemic toxicity.The syntheses and exceptional antitumor activities of a series of novel fatty acid-second-generation taxoid conjugates against human tumor xenografts are reported.
Co-reporter:Bruno D. Chapsal, Zihao Hua, Iwao Ojima
Tetrahedron: Asymmetry 2006 Volume 17(Issue 4) pp:642-657
Publication Date(Web):20 February 2006
DOI:10.1016/j.tetasy.2005.12.035
A library of new fine-tunable monodentate phosphite and phosphoramidite ligands based on chiral biphenol have been designed and developed. These monodentate phosphorus ligands have exhibited excellent enantioselectivity in the Pd-catalyzed asymmetric allylic alkylation and Rh-catalyzed asymmetric hydrogenation.N,N-[(S)-1-(Naphthalen-2-yl)ethyl][(S)-1-(2-methoxyphenyl)ethyl]amineC21H23NOEe: 99% (by 1H NMR of the corresponding diastereoisomer)[α]D22=-187 (c 1.66, CHCl3)Source of chirality: (S)-(−)-1-(2-naphthyl)ethylamine (99.0%)Absolute configuration: (S,S)O,O′-(S)-(5,5′,6,6′-Tetramethyl-2,2′-diyl)-N,N-bis[(S)-1-(naphthalen-1-yl)ethyl]phosphoramiditeC40H38NO2PEe: 99% (by 1H NMR of the corresponding diastereoisomer)[α]D22=+168.2 (c 1.32, CHCl3)Source of chirality: resolutionAbsolute configuration: (S,S,S)O,O′-(S)-(5,5′,6,6′-Tetramethyl-2,2′-diyl)-N,N-bis[(S)-1-(naphthalen-2-yl)ethyl]phosphoramiditeC40H38NO2PEe: 99% (by 1H NMR of the corresponding diastereoisomer)[α]D22=-415.3 (c 0.98, CHCl3)Source of chirality: resolutionAbsolute configuration: (S,S,S)O,O′-(S)-(5,5′,6,6′-Tetramethyl-2,2′-diyl)-N,N-[(S)-1-phenylethyl][(S)-1-(2-methoxyphenyl)ethyl]phosphoramiditeC33H37NO3PEe: 99% (by 1H NMR of the corresponding diastereoisomer)[α]D22=-102.4 (c 1.25, CHCl3)Source of chirality: resolutionAbsolute configuration: (S,S,S)O,O′-(S)-(5,5′,6,6′-Tetramethyl-2,2′-diyl)-N,N-[(S)-1-phenylethyl][(S)-1-(2-methylphenyl)ethyl]phosphoramiditeC33H36NO2PEe: 99% (by 1H NMR of the corresponding diastereoisomer)[α]D22=-126 (c 1.10, CHCl3)Source of chirality: resolutionAbsolute configuration: (S,S,S)O,O′-(S)-(5,5′,6,6′-Tetramethyl-2,2′-diyl)-N,N-bis[(S)-1-(2-methylphenyl)ethyl]phosphoramiditeC34H39NO2PEe: 99% (by 1H NMR of the corresponding diastereoisomer)[α]D22=+72 (c 1.05, CHCl3)Source of chirality: resolutionAbsolute configuration: (S,S,S)O,O′-(S)-(5,5′,6,6′-Tetramethyl-2,2′-diyl)-N,N-bis[(S)-1-(2-methoxyphenyl)ethyl]phosphoramiditeC34H39NO3PEe: 99% (by 1H NMR of the corresponding diastereoisomer)[α]D22=+34.4 (c 0.64, CHCl3)Source of chirality: resolutionAbsolute configuration: (S,S,S)O,O′-(S)-(5,5′,6,6′-Tetramethyl-2,2′-diyl)-N,N-[(S)-1-(naphthalen-1-yl)ethyl][(S)-1-(2-methoxyphenyl)ethyl]phosphoramiditeC37H39NO3PEe: 99% (by 1H NMR of the corresponding diastereoisomer)[α]D22=+96.7 (c 1.23, CHCl3)Source of chirality: resolutionAbsolute configuration: (S,S,S)O,O′-(S)-(5,5′,6,6′-Tetramethyl-2,2′-diyl)-N,N-[(S)-1-(naphthalen-2-yl)ethyl][(S)-1-(2-methoxyphenyl)ethyl]phosphoramiditeC37H39NO3PEe: 99% (by 1H NMR of the corresponding diastereoisomer)[α]D22=+246 (c 0.80, CHCl3)Source of chirality: resolutionAbsolute configuration: (S,S,S)(3aR,7aS)-1-[6-Bromo-3,4-(methylenedioxy)benzyl]-3a,4,5,7a-tetrahydroindolin-2-oneC17H22NOEe: 99.4% (by HPLC Chiralpak ADRH CH3CN/H2O 60:40)[α]D22=-40.0 (c 0.5, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (R,S)(3aR,11bS,11cS)-4-Methoxycarbonyl-3,3a,4,7,11b,11c-hexahydro-9,10-(methylenedioxy)pyrrolo[3,2,1de]phenanthridin-5- oneC18H18NO5Ee: 99.4% (by 1H NMR of the corresponding diastereoisomer)[α]D22=-19.6 (c 0.51, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (R,S,S)(3aR,12bS,12cS)-5-Oxo-γ-lycoraneC16H16NO3PEe: 99.4% (by 1H NMR of the corresponding diastereoisomer)[α]D22=+83.8 (c 0.74, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (R,S,S)(+)-γ-LycoraneC16H20NO2Ee: 99.4% (by 1H NMR of the corresponding diastereoisomer)[α]D22=+18.1 (c 1.10, EtOH)Source of chirality: asymmetric synthesisAbsolute configuration: (S,S,S)
Co-reporter:Xudong Geng, Raphaël Geney, Paula Pera, Ralph J. Bernacki, Iwao Ojima
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 13) pp:3491-3494
Publication Date(Web):5 July 2004
DOI:10.1016/j.bmcl.2004.04.060
Based on a common pharmacophore model and the hypothesis that the baccatin core of taxoids is a scaffold securing the proper orientation of the side chains, a bicyclic alkaloid scaffold was designed as a baccatin surrogate. Using this scaffold, two novel macrocyclic and open-chain ‘taxoid-mimicking’ compounds were synthesized. Two of these ‘taxoid-mimics’, 2 and 3, were found to possess cytotoxicity with micromolar level IC50 values against human breast cancer cell lines.The design, synthesis, and biological activity of several novel cytotoxic alkaloids, mimicking and simplifying the taxoid skeleton are reported.
Co-reporter:Zihao Hua;Victor C. Vassar;Hojae Choi
PNAS 2004 Volume 101 (Issue 15 ) pp:5411-5416
Publication Date(Web):2004-04-13
DOI:10.1073/pnas.0307101101
Monodentate phosphoramidite ligands have been developed based on enantiopure 6,6′-dimethylbiphenols with axial chirality.
These chiral ligands are easy to prepare and flexible for modifications. The fine-tuning capability of these ligands plays
a significant role in achieving high enantioselectivity in the asymmetric hydroformylation of allyl cyanide and the conjugate
addition of diethylzinc to cycloalkenones.
Co-reporter:Iwao Ojima
ChemBioChem 2004 Volume 5(Issue 5) pp:
Publication Date(Web):28 APR 2004
DOI:10.1002/cbic.200300844
Flurinated prodrugs. This minireview describes the exploitation of the unique nature of fluorine in the medicinal chemistry and chemical biology of taxane anticancer agents as a showcase in this field of research.
Co-reporter:Iwao Ojima, Cecilia L Fumero-Oderda, Scott D Kuduk, Zhuping Ma, Fumiko Kirikae, Teruo Kirikae
Bioorganic & Medicinal Chemistry 2003 Volume 11(Issue 13) pp:2867-2888
Publication Date(Web):3 July 2003
DOI:10.1016/S0968-0896(03)00181-0
A series of new taxoids modified at the C-3′, C-3′N, C-10, C-2 and C-7 positions has been designed, synthesized and evaluated for their potency to induce NO and TNF production by peritoneal murine macrophages (Mφ) from LPS-responsive C3H/HeN and LPS-hyporesponsive C3H/HeJ strains and human blood cells, and for their ability to inhibit the growth of Mφ-like cell lines J774.1 and J7.DEF3. The SAR-study has shown that the nature of the substituents at these positions have critical effect on the induction of TNF and NO production by Mφ. Positions C-3′ and C-10 are the most flexible and an intriguing effect of the length of the substituents at the C-10 position is observed for taxoids bearing a straight chain alkanoyl moiety. An aromatic group at the C-3′N and C-2 positions is required for the activity, while only hydroxyl or acetyl substituents seem to be tolerated at the C-7 position. The natural stereochemistry in the C-13 isoserine side chain of the taxoids is an absolute requirement for macrophage activation. It has also been clearly shown that there is no correlation between the ability of the taxoids to induce TNF/NO production in C3H/HeN Mφ and the cytotoxicity against Mφ-like cells.A series of new taxoids modified at the C-3′, C-3′N, C-10, C-2 and C-7 positions are designed, synthesized and evaluated for their potency to induce NO and TNF production by peritoneal murine macrophages (Mφ) and for their ability to inhibit the growth of Mφ-like cell lines.
Co-reporter:Iwao Ojima, Xudong Geng, Songnian Lin, Paula Pera, Ralph J Bernacki
Bioorganic & Medicinal Chemistry Letters 2002 Volume 12(Issue 3) pp:349-352
Publication Date(Web):11 February 2002
DOI:10.1016/S0960-894X(01)00747-8
A series of novel macrocyclic taxoids was designed and synthesized by connecting the C-2 and C-3′ N positions of the taxoid framework with various tethers. Cytotoxicity of these macrocyclic taxoids was evaluated against a human breast cancer cell line LCC6-WT, and a couple of the taxoids exhibited 0.09–0.3 μM IC50 values.Novel cytotoxic macrocyclic taxoids with various linkers connecting the C2 and C3′ N positions of taxoid framework are reported.
Co-reporter:Victor C. Vassar;Chih-Yuan Chuang;Zuping Ma;Raphaël Geney
Chirality 2002 Volume 14(Issue 2‐3) pp:151-162
Publication Date(Web):1 FEB 2002
DOI:10.1002/chir.10050
Regio- and enantioselectivity in the asymmetric aminohydroxylation (AA) reaction of O-substituted 4-hydroxy-2-butenoates as well as the mechanism of the reaction were studied. When the electronic properties of the phenyl group in a substrate were altered by using different substituents, two conflicting trends were observed: The O-benzoyl substrates showed greater regio- and enantioselectivity when an electron-donating substituent was attached at the C-4 position of the phenyl group, while the O-benzyl substrates exhibited better regio- and enantioselectivity with an electron-withdrawing substituent at the C-4 position of the phenyl moiety. Thus, these results have disclosed hitherto unknown remarkable electronic effects in the AA reaction. Detailed analysis of possible electronic interactions in the chiral catalyst–substrate complex has revealed the importance of dipolar aromatic–aromatic interactions between the aromatic substituent of the substrate and the nitrogen heteroaromatic moiety of the chiral ligand for effective regiocontrol as well as enantioface selection in the AA reaction. A plausible model of the key intermediate in the AA reaction of O-substituted 4-hydroxy-2-butenoates is proposed. Chirality 14:151–162, 2002. © 2002 Wiley-Liss, Inc.
Co-reporter:Michael L. Miller
The Chemical Record 2001 Volume 1(Issue 3) pp:
Publication Date(Web):18 JUN 2001
DOI:10.1002/tcr.1008
Taxol® (paclitaxel) and Taxotère® (docetaxel) are currently considered to be among the most important anticancer drugs in cancer chemotherapy. The anticancer activity of these drugs is ascribed to their unique mechanism of action, i.e., causing mitotic arrest in cancer cells, leading to apoptosis through inhibition of the depolymerization of microtubules. Although both paclitaxel and docetaxel possess potent antitumor activity, treatment with these drugs often results in a number of undesirable side effects, as well as multidrug resistance (MDR). Therefore, it has become essential to develop new anticancer agents with superior pharmacological properties, improved activity against various classes of tumors, and fewer side effects. This paper describes an account of our research on the chemistry of paclitaxel and taxoid anticancer agents at the biomedical interface, including:
- 1
The structure-activity relationship (SAR) study of taxoids leading to the development of the “second-generation” taxoids, which possess exceptional activity against drug-resistant cancer cells expressing the MDR phenotype.
- 2
Development of fluorinated taxoids to study the bioactive conformation of paclitaxel and photoaffinity labeling taxoids for mapping of the drug-binding domain on both microtubules and P-glycoprotein.
- 3
The synthesis of novel macrocyclic taxoids for the investigation into the common pharmacophore for microtubule stabilizing anticancer agents.
© 2001 John Wiley & Sons, Inc. and The Japan Chemical Journal Forum Chem Rec 1:195–211, 2001
Co-reporter:Iwao Ojima, Songnian Lin, John C Slater, Tao Wang, Paula Pera, Ralph J Bernacki, Cristiano Ferlini, Giovanni Scambia
Bioorganic & Medicinal Chemistry 2000 Volume 8(Issue 7) pp:1619-1628
Publication Date(Web):July 2000
DOI:10.1016/S0968-0896(00)00093-6
A series of new taxoids bearing difluoromethyl group at the C-3′ position and modifications at the C-10 and C-14 positions has been synthesized and their biological activities studied. The in vitro cytotoxicity assay results indicate that these newly developed taxoids exhibit comparable to several times better activity against drug-sensitive cell line LCC6-WT, and 40–70 times better activity against the corresponding drug-resistant cancer cell line LCC6-MDR as compared to that of paclitaxel. Apoptosis analysis has revealed the exceptional activity of SB-T-12843 (1e) in inducing apoptosis in both MDR-bearing and MDR-negative cancer cells.
Co-reporter:Songnian Lin;Xudong Geng;David J. Gallagher;Chuanxing Qu;Robert Tynebor;Jessica Rutter;Elizabeth Pollina
Chirality 2000 Volume 12(Issue 5‐6) pp:431-441
Publication Date(Web):19 MAY 2000
DOI:10.1002/(SICI)1520-636X(2000)12:5/6<431::AID-CHIR24>3.0.CO;2-M
A series of highly potent second-generation taxoids bearing a 2-methylprop-1-enyl or a 2-methylpropyl group at C-3′ with modifications at the C-2, C-10, and C-14 positions was synthesized through the coupling of racemic cis-β-lactams with properly protected/modified baccatin and 14-OH-baccatin. A high level of kinetic resolution was observed for all cases examined. The observed highly efficient enantiomer differentiation is ascribed to the markedly different chiral environment between the (+)- and (−)-β-lactams in their approach to the chiral framework of the enantiopure lithium alkoxide of a baccatin in the ring-opening coupling process. It was also observed that substantially higher selectivity was achieved when 14-OH-baccatin-1,14-carbonate was used. Analysis of the transition state models revealed that the repulsive interactions between the 3-TIPS group of a (−)-β-lactam with 1,14-carbonate group of the baccatin substantially increases the asymmetric bias in the kinetic resolution process, favoring the reaction of a (+)-β-lactam, which leads to the observed excellent selectivity. Chirality 12:431–441, 2000. © 2000 Wiley-Liss, Inc.
Co-reporter:Joseph J. Kaloko, Yu-Han Gary Teng and Iwao Ojima
Chemical Communications 2009(Issue 30) pp:NaN4571-4571
Publication Date(Web):2009/06/29
DOI:10.1039/B909781C
Rapid construction of 5-7-6-5 fused tetracyclic carbocycles and heterocycles from cyclohexene-diynes and CO has been achieved in one step through a Rh(I)-catalyzed [2 + 2 + 2 + 1] cycloaddition process.
.jpg)












![3-Methyl-2-oxo-2,3-dihydrobenzo[d]oxazole-6-sulfonyl chloride](http://img.cochemist.com/ccimg/62600/62522-63-8.png)
![3-Methyl-2-oxo-2,3-dihydrobenzo[d]oxazole-6-sulfonyl chloride](http://img.cochemist.com/ccimg/62600/62522-63-8_b.png)










![Ethane, 1-azido-2-[2-[2-(2-fluoroethoxy)ethoxy]ethoxy]-](/data/chemimg/3722800/1233816-91-5.png)
![Ethane, 1-azido-2-[2-[2-(2-fluoroethoxy)ethoxy]ethoxy]-](/data/chemimg/3722800/1233816-91-5_b.png)
![Butanoic acid, 4-[[2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]ethyl]amino]-4-oxo-](/data/chemimg/3722900/1202400-17-6.png)
![Butanoic acid, 4-[[2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]ethyl]amino]-4-oxo-](/data/chemimg/3722900/1202400-17-6_b.png)




![2-Methyl-2-propanyl 2-[({[(2-methyl-2-propanyl)oxy]carbonyl}oxy)methyl]-2-propen-1-yl carbonate](http://img.cochemist.com/ccimg/620200/620161-75-3.png)
![2-Methyl-2-propanyl 2-[({[(2-methyl-2-propanyl)oxy]carbonyl}oxy)methyl]-2-propen-1-yl carbonate](http://img.cochemist.com/ccimg/620200/620161-75-3_b.png)






![5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide](http://img.cochemist.com/ccimg/192800/192703-06-3.png)
![5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide](http://img.cochemist.com/ccimg/192800/192703-06-3_b.png)




![Benzo[b]thiophen-2(3H)-one, 5-fluoro-](http://img.cochemist.com/ccimg/125800/125772-32-9.png)
![Benzo[b]thiophen-2(3H)-one, 5-fluoro-](http://img.cochemist.com/ccimg/125800/125772-32-9_b.png)
![Spiro[1,3-dioxolane-2,6'(5'H)-quinoline], 7',8'-dihydro-2'-methoxy-](http://img.cochemist.com/ccimg/120700/120685-99-6.png)
![Spiro[1,3-dioxolane-2,6'(5'H)-quinoline], 7',8'-dihydro-2'-methoxy-](http://img.cochemist.com/ccimg/120700/120685-99-6_b.png)
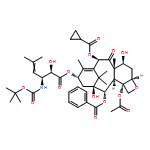
![Benzenepropanoic acid, b-(benzoylamino)-a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-,(2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-6,12b-bis(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-4,11-dihydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-ylester, (aR,bS)-](http://img.cochemist.com/ccimg/114700/114655-02-6.png)
![Benzenepropanoic acid, b-(benzoylamino)-a-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-,(2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-6,12b-bis(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-4,11-dihydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-ylester, (aR,bS)-](http://img.cochemist.com/ccimg/114700/114655-02-6_b.png)
![2,5-Pyrrolidinedione, 1-[(propoxycarbonyl)oxy]-](http://img.cochemist.com/ccimg/112900/112884-31-8.png)
![2,5-Pyrrolidinedione, 1-[(propoxycarbonyl)oxy]-](http://img.cochemist.com/ccimg/112900/112884-31-8_b.png)






![5-FLUOROBENZO[B]THIOPHENE](http://img.cochemist.com/ccimg/70100/70060-12-7.png)
![5-FLUOROBENZO[B]THIOPHENE](http://img.cochemist.com/ccimg/70100/70060-12-7_b.png)
![2,5-Pyrrolidinedione, 1-[(butoxycarbonyl)oxy]-](http://img.cochemist.com/ccimg/64600/64562-12-5.png)
![2,5-Pyrrolidinedione, 1-[(butoxycarbonyl)oxy]-](http://img.cochemist.com/ccimg/64600/64562-12-5_b.png)













![1,3-Dioxolo[4,5-g]quinazolin-6,8(5H,7H)-dione](http://img.cochemist.com/ccimg/21900/21884-35-5.png)
![1,3-Dioxolo[4,5-g]quinazolin-6,8(5H,7H)-dione](http://img.cochemist.com/ccimg/21900/21884-35-5_b.png)






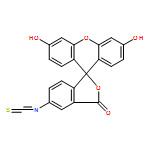
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)
![Octadecanamide,N-[(1S,2R)-2-hydroxy-1-(hydroxymethyl)heptadecyl]-](http://img.cochemist.com/ccimg/2400/2304-80-5.png)
![Octadecanamide,N-[(1S,2R)-2-hydroxy-1-(hydroxymethyl)heptadecyl]-](http://img.cochemist.com/ccimg/2400/2304-80-5_b.png)
















![1,3-Benzenediol,2-[(1R,6R)-3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-5-pentyl-](http://img.cochemist.com/ccimg/14000/13956-29-1.png)
![1,3-Benzenediol,2-[(1R,6R)-3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-5-pentyl-](http://img.cochemist.com/ccimg/14000/13956-29-1_b.png)





![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7.png)
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7_b.png)