Co-reporter:Zachary Miller; Keun-Sik Kim; Do-Min Lee; Vinod Kasam; Si Eun Baek; Kwang Hyun Lee; Yan-Yan Zhang; Lin Ao; Kimberly Carmony; Na-Ra Lee; Shou Zhou; Qingquan Zhao; Yujin Jang; Hyun-Young Jeong; Chang-Guo Zhan; Wooin Lee; Dong-Eun Kim
Journal of Medicinal Chemistry 2015 Volume 58(Issue 4) pp:2036-2041
Publication Date(Web):February 6, 2015
DOI:10.1021/jm501344n
We performed a virtual screen of ∼340 000 small molecules against the active site of proteasomes followed by in vitro assays and subsequent optimization, yielding a proteasome inhibitor with pyrazole scaffold. The pyrazole-scaffold compound displayed excellent metabolic stability and was highly effective in suppressing solid tumor growth in vivo. Furthermore, the effectiveness of this compound was not negatively impacted by resistance to bortezomib or carfilzomib.
Co-reporter:Kimberly Cornish Carmony;Dr. Lalit Kumar Sharma;Do-Min Lee;Ji Eun Park;Dr. Wooin Lee;Dr. Kyung-Bo Kim
ChemBioChem 2015 Volume 16( Issue 2) pp:284-292
Publication Date(Web):
DOI:10.1002/cbic.201402491
Abstract
In addition to two well-recognized proteasome subtypes—constitutive proteasomes and immunoproteasomes—mounting evidence also suggests the existence of intermediate proteasome subtypes containing unconventional mixtures of catalytic subunits. Although they appear to play unique biological roles, the lack of practical methods for detecting distinct proteasome subtypes has limited functional investigations. Here, we report the development of activity-based probes that crosslink two catalytic subunits within intact proteasome complexes. Identification of the crosslinked subunit pairs provides direct evidence of the catalytic subunit composition of proteasomes. Using these probes, we found that U266 multiple myeloma cells contain intermediate proteasomes comprising both β1i and β2, but not β1 and β2i, consistent with previous findings with other cell types. Our bifunctional probes can be utilized in functional investigations of distinct proteasome subtypes in various biological settings.
Co-reporter:Ji Eun Park, Ying Wu, Kimberly Cornish Carmony, Zachary Miller, Lalit Kumar Sharma, Do-Min Lee, Doo-Young Kim, Wooin Lee and Kyung-Bo Kim
Molecular BioSystems 2014 vol. 10(Issue 2) pp:196-200
Publication Date(Web):14 Nov 2013
DOI:10.1039/C3MB70471H
Mammalian cells have two main types of proteasomes, the constitutive proteasome and the immunoproteasome, each containing a distinct set of three catalytic subunits. Recently, additional proteasome subtypes containing a non-standard mixture of catalytic subunits have gained increasing attention, especially due to their presence in cancer settings. However, practical methods for identifying proteasome subtypes have been lacking. Here, we report the development of the first fluorescence resonance energy transfer (FRET)-based strategy that can be utilized to identify different proteasome subtypes present within cells. We have developed FRET donor- and acceptor-probes that are based on previously reported peptide epoxyketones and selectively target individual proteasome catalytic subunits. Using the purified proteasome and cancer cell lysates, we demonstrate the feasibility of a FRET-based approach for determining the catalytic subunit composition of individual 20S proteasome subtypes. Ultimately, this approach may be utilized to study the functions of individual proteasome subtypes in cells.
Co-reporter:Kimberly Cornish Carmony
Cell Biochemistry and Biophysics 2013 Volume 67( Issue 1) pp:91-101
Publication Date(Web):2013 September
DOI:10.1007/s12013-013-9626-4
Over the years, the proteasome has been extensively investigated due to its crucial roles in many important signaling pathways and its implications in diseases. Two proteasome inhibitors—bortezomib and carfilzomib—have received FDA approval for the treatment of multiple myeloma, thereby validating the proteasome as a chemotherapeutic target. As a result, further research efforts have been focused on dissecting the complex biology of the proteasome to gain the insight required for developing next-generation proteasome inhibitors. It is clear that chemical probes have made significant contributions to these efforts, mostly by functioning as inhibitors that selectively block the catalytic activity of proteasomes. Analogues of these inhibitors are now providing additional tools for visualization of catalytically active proteasome subunits, several of which allow real-time monitoring of proteasome activity in living cells as well as in in vivo settings. These imaging probes will provide powerful tools for assessing the efficacy of proteasome inhibitors in clinical settings. In this review, we will focus on the recent efforts towards developing imaging probes of proteasomes, including the latest developments in immunoproteasome-selective imaging probes.
Co-reporter:Kimberly Cornish Carmony, Do-Min Lee, Ying Wu, Na-Ra Lee, Marie Wehenkel, Jason Lee, Beilei Lei, Chang-Guo Zhan, Kyung-Bo Kim
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 2) pp:607-613
Publication Date(Web):15 January 2012
DOI:10.1016/j.bmc.2011.06.039
While the constitutive, 26S proteasome plays an important role in regulating many important cellular processes, a variant form known as the immunoproteasome is thought to primarily function in adaptive immune responses. However, recent studies indicate an association of immunoproteasomes with many physiological disorders such as cancer, neurodegenerative, and inflammatory diseases. Despite this, the detailed functions of the immunoproteasome remain poorly understood. Immunoproteasome-specific probes are essential to gain insight into immunoproteasome function. Here, we describe for the first time the development of cell-permeable activity-based fluorescent probes, UK101-Fluor and UK101-B660, which selectively target the catalytically active LMP2/β1i subunit of the immunoproteasome. These probes facilitate rapid detection of the cellular localization of catalytically active immunoproteasomes in living cells, providing a valuable tool to analyze immunoproteasome functions. Additionally, as LMP2/β1i may serve as a potential tumor biomarker, an LMP2/β1i-targeting fluorescent imaging probe may be applicable to a rapid readout assay to determine tumor LMP2/β1i levels.
Co-reporter:Kedra Cyrus, Marie Wehenkel, Eun-Young Choi, Hyeong-Jun Han, Hyosung Lee, Hollie Swanson and Kyung-Bo Kim
Molecular BioSystems 2011 vol. 7(Issue 2) pp:359-364
Publication Date(Web):04 Oct 2010
DOI:10.1039/C0MB00074D
Conventional genetic approaches have provided a powerful tool in the study of proteins. However, these techniques often preclude selective manipulation of temporal and spatial protein functions, which is crucial for the investigation of dynamic cellular processes. To overcome these limitations, a small molecule-based novel technology termed “PROteolysis TArgeting ChimeraS (PROTACs)” has been developed, targeting proteins for degradation at the post-translational level. Despite the promising potential of PROTACs to serve as molecular probes of complex signaling pathways, their design has not been generalized for broad application. Here, we present the first generalized approach for PROTAC design by fine-tuning the distance between the two participating partner proteins, the E3 ubiquitin ligase and the target protein. As such, we took a chemical approach to create estrogen receptor (ER)-α targeting PROTACs with varying linker lengths and the loss of the ER in cultured cells was monitored via western blot and fluorometric analyses. We found a significant effect of chain length on PROTAC efficacy, and, in this case, the optimum distance between the E3 recognition motif and the ligand was a 16 atom chain length. The information gathered from this experiment may offer a generalizable PROTAC design strategy to further the expansion of the PROTAC toolbox, opening new possibilities for the broad application of the PROTAC strategy in the study of multiple signaling pathways.
Co-reporter:Beilei Lei, Mohamed Diwan M. Abdul Hameed, Adel Hamza, Marie Wehenkel, Jennifer L. Muzyka, Xiao-Jun Yao, Kyung-Bo Kim, and Chang-Guo Zhan
The Journal of Physical Chemistry B 2010 Volume 114(Issue 38) pp:12333-12339
Publication Date(Web):September 2, 2010
DOI:10.1021/jp1058098
Given that immunoproteasome inhibitors are currently being developed for a variety of potent therapeutic purposes, the unique specificity of an α′,β′-epoxyketone peptide (UK101) toward the LMP2 subunit of the immunoproteasome (analogous to β5 subunit of the constitutive proteasome) has been investigated in this study for the first time by employing homology modeling, molecular docking, molecular dynamics simulation, and molecular mechanics Poisson−Boltzmann surface area (MM-PBSA) binding free energy calculations. On the basis of the simulated binding structures, the calculated binding free energies are in qualitative agreement with the corresponding experimental data, and the selectivity of UK101 is explained reasonably. The observed selectivity of UK101 for the LMP2 subunit is rationalized by the requirement for both a linear hydrocarbon chain at the N terminus and a bulky group at the C terminus of the inhibitor, because the LMP2 subunit has a much more favorable hydrophobic pocket interacting with the linear hydrocarbon chain, and the bulky group at the C terminus has a steric clash with the Tyr 169 in β5 subunit. Finally, our results help to clarify why UK101 is specific to the LMP2 subunit of immunoproteasome, and this investigation should be valuable for rational design of more potent LMP2-specific inhibitors.
Co-reporter:Kedra Cyrus Dr.;Marie Wehenkel;Eun-Young Choi Dr.;Hyosung Lee Dr.;Hollie Swanson Dr.;Kyung-Bo Kim Dr.
ChemMedChem 2010 Volume 5( Issue 7) pp:979-985
Publication Date(Web):
DOI:10.1002/cmdc.201000146
Abstract
Estrogen receptor-α (ER) antagonists have been widely used for breast cancer therapy. Despite initial responsiveness, hormone-sensitive ER-positive cancer cells eventually develop resistance to ER antagonists. It has been shown that in most of these resistant tumor cells, the ER is expressed and continues to regulate tumor growth. Recent studies indicate that tamoxifen initially acts as an antagonist, but later functions as an ER agonist, promoting tumor growth. This suggests that targeted ER degradation may provide an effective therapeutic approach for breast cancers, even those that are resistant to conventional therapies. With this in mind, we previously demonstrated that proteolysis targeting chimeras (PROTACs) effectively induce degradation of the ER as a proof-of-concept experiment. Herein we further refined the PROTAC approach to target the ER for degradation. The ER-targeting PROTACs are composed of an estradiol on one end and a hypoxia-inducing factor 1α (HIF-1α)-derived synthetic pentapeptide on the other. The pentapeptide is recognized by an E3 ubiquitin ligase called the von Hippel Lindau tumor suppressor protein (pVHL), thereby recruiting the ER to this E3 ligase for ubiquitination and degradation. Specifically, the pentapeptide is attached at three different locations on estradiol to generate three different PROTAC types. With the pentapeptide linked through the C7α position of estradiol, the resulting PROTAC shows the most effective ER degradation and highest affinity for the estrogen receptor. This result provides an opportunity to develop a novel type of ER antagonist that may overcome the resistance of breast tumors to conventional drugs such as tamoxifen and fulvestrant (Faslodex).
Co-reporter:Kyung Bo Kim ;Craig M. Crews
Journal of Medicinal Chemistry 2008 Volume 51(Issue 9) pp:2600-2605
Publication Date(Web):April 5, 2008
DOI:10.1021/jm070421s
Co-reporter:Marie Wehenkel, Jin Tae Hong and Kyung Bo Kim
Molecular BioSystems 2008 vol. 4(Issue 4) pp:280-286
Publication Date(Web):20 Feb 2008
DOI:10.1039/B716221A
Primarily used for medicinal purposes in the past, biologically active small molecules have been increasingly employed to explore complex biological processes in the era of “chemical genetics”. Since the contributions of this small molecule approach to biology have been extensive, we limit the focus of our review to the use of small-molecule modulators in the exciting field of proteasomal biology, one that has benefited significantly from a chemical genetics approach. Specifically, as the contributions of general inhibitors of proteasomal activity to the fields of cell biology and clinical oncology have been extensively discussed in several excellent reviews, we instead outline recent progress towards the development of novel, specific classes of proteasome modulators for studies of proteasomal biology and the types of proteasome inhibitors emerging as important new treatment options for cancer therapeutics.
Co-reporter:Yik Khuan (Abby) Ho, Paola Bargagna-Mohan, Marie Wehenkel, Royce Mohan, Kyung-Bo Kim
Chemistry & Biology 2007 Volume 14(Issue 4) pp:419-430
Publication Date(Web):20 April 2007
DOI:10.1016/j.chembiol.2007.03.008
The immunoproteasome, having been linked to neurodegenerative diseases and hematological cancers, has been shown to play an important role in MHC class I antigen presentation. However, its other pathophysiological functions are still not very well understood. This can be attributed mainly to a lack of appropriate molecular probes that can selectively modulate the immunoproteasome catalytic subunits. Herein, we report the development of molecular probes that selectively inhibit the major catalytic subunit, LMP2, of the immunoproteasome. We show that these compounds irreversibly modify the LMP2 subunit with high specificity. Importantly, LMP2-rich cancer cells compared to LMP2-deficient cancer cells are more sensitive to growth inhibition by the LMP2-specific inhibitor, implicating an important role of LMP2 in regulating cell growth of malignant tumors that highly express LMP2.
Co-reporter:Hyosung Lee;Dinesh Puppala;Eun-Young Choi Dr.;Hollie Swanson Dr.;Kyung-Bo Kim Dr.
ChemBioChem 2007 Volume 8(Issue 17) pp:
Publication Date(Web):28 SEP 2007
DOI:10.1002/cbic.200700438
License to degrade. A chimeric small molecule or PROTAC (PROteolysis TArgeting Chimera) based on the chemopreventive natural product apigenin was developed (see scheme). The molecule was shown to target the aryl hydrocarbon receptor (AHR) for degradation by the proteasome.

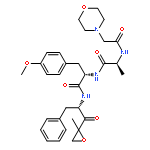
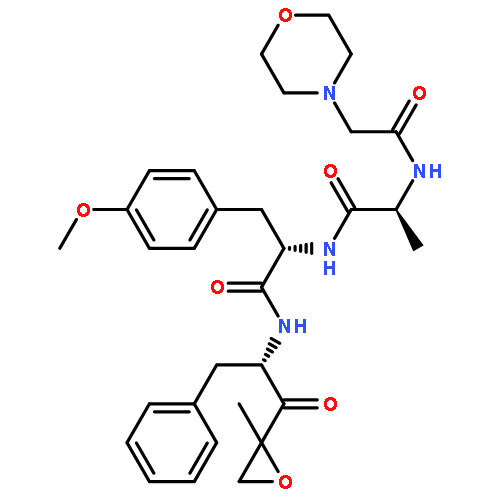
![L-Threoninamide,N-acetyl-N-methyl-L-isoleucyl-L-isoleucyl-N-[(1S)-3-methyl-1-[[(2R)-2-methyl-2-oxiranyl]carbonyl]butyl]-](http://img.cochemist.com/ccimg/134400/134381-21-8.png)
![L-Threoninamide,N-acetyl-N-methyl-L-isoleucyl-L-isoleucyl-N-[(1S)-3-methyl-1-[[(2R)-2-methyl-2-oxiranyl]carbonyl]butyl]-](http://img.cochemist.com/ccimg/134400/134381-21-8_b.png)