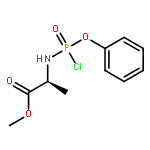
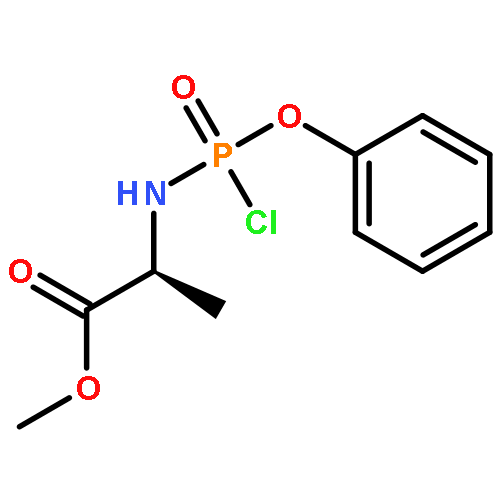
Co-reporter:Nadège Hamon, Magdalena Slusarczyk, Michaela Serpi, Jan Balzarini, Christopher McGuigan
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 4) pp:829-838
Publication Date(Web):15 February 2015
DOI:10.1016/j.bmc.2014.12.039
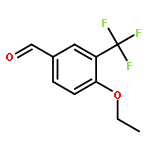
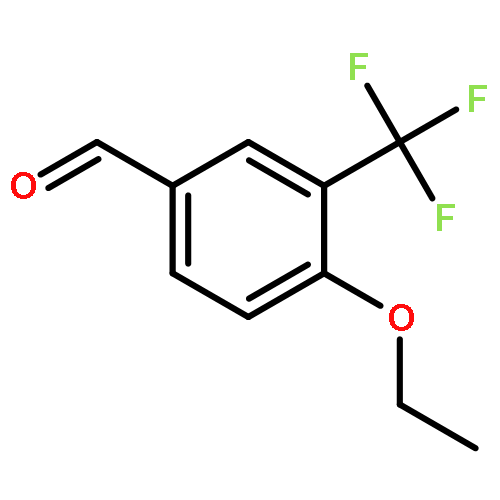
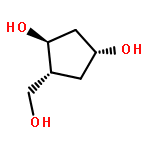
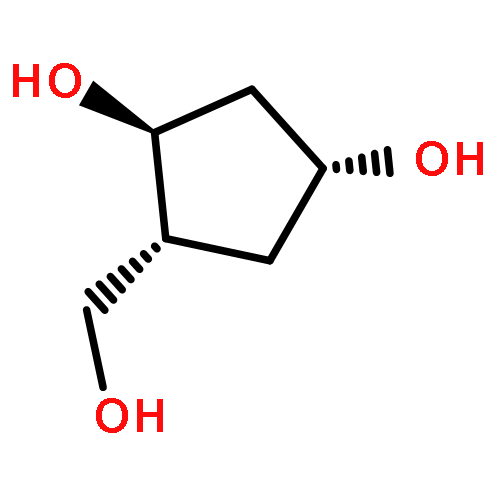
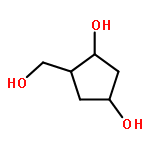
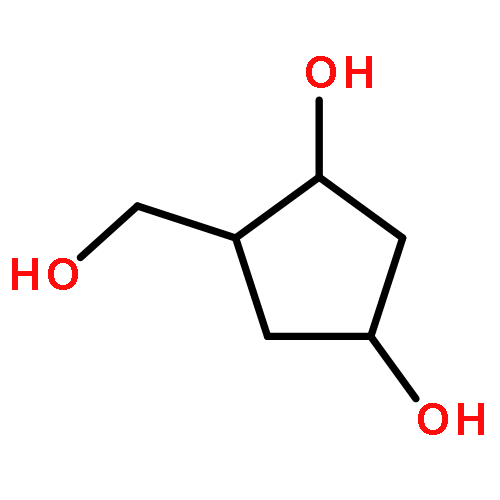
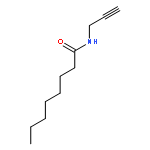
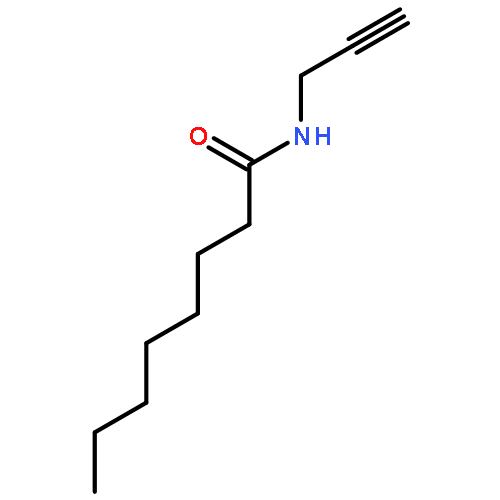
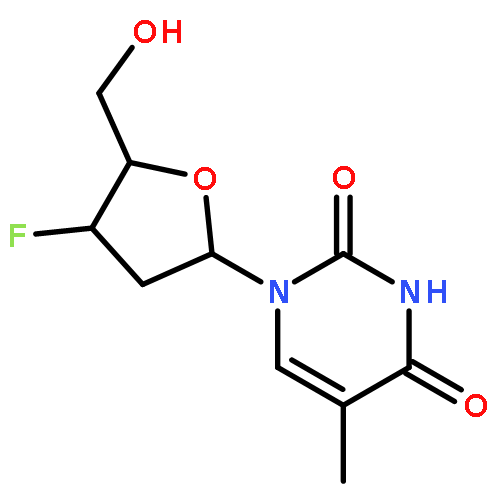
2-Deoxy-α-d-ribose-1-phosphate is of great interest as it is involved in the biosynthesis and/or catabolic degradation of several nucleoside analogues of biological and therapeutic relevance. However due to the lack of a stabilising group at its 2-position, it is difficult to synthesize stable prodrugs of this compound. In order to overcome this lack of stability, the synthesis of carbasugar analogues of 2-deoxyribose-1-phosphate was envisioned. Herein the preparation of a series of prodrugs of two carbocyclic analogues of 2-deoxyribose-1-phosphate using the phosphoramidate ProTide technology, along with their biological evaluation against HIV and cancer cell proliferation, is reported.
Co-reporter:Magdalena Slusarczyk ; Monica Huerta Lopez ; Jan Balzarini ; Malcolm Mason ; Wen G. Jiang ; Sarah Blagden ; Emely Thompson ; Essam Ghazaly
Journal of Medicinal Chemistry 2014 Volume 57(Issue 4) pp:1531-1542
Publication Date(Web):January 28, 2014
DOI:10.1021/jm401853a
Gemcitabine is a nucleoside analogue commonly used in cancer therapy but with limited efficacy due to a high susceptibility to cancer cell resistance. The addition of a phosphoramidate motif to the gemcitabine can protect it against many of the key cancer resistance mechanisms. We have synthesized a series of gemcitabine phosphoramidate prodrugs and screened for cytostatic activity in a range of different tumor cell lines. Among the synthesized compounds, one in particular (NUC-1031, 6f) was shown to be potent in vitro. Importantly, compared with gemcitabine, 6f activation was significantly less dependent on deoxycytidine kinase and on nucleoside transporters, and it was resistant to cytidine deaminase-mediated degradation. Moreover, 6f showed a significant reduction in tumor volumes in vivo in pancreatic cancer xenografts. The ProTide 6f is now in clinical development with encouraging efficacy signals in a Phase I/II study, which strongly supports the ProTide approach to generate promising new anticancer agents.
Co-reporter:Nadege Hamon, Maurizio Quintiliani, Jan Balzarini, Christopher McGuigan
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 9) pp:2555-2559
Publication Date(Web):1 May 2013
DOI:10.1016/j.bmcl.2013.02.117
We report in this Letter the synthesis of prodrugs of 2-fluoro-2-deoxyarabinose-1-phosphate and 2,2-difluoro-2-deoxyribose-1-phosphate. We demonstrate the difficulty of realising a phosphorylation step on the anomeric position of 2-deoxyribose, and we discover that introduction of fluorine atoms on the 2 position of 2-deoxyribose enables the phosphorylation step: in fact, the stability of the prodrugs increases with the degree of 2-fluorination. Stability studies of produgs of 2-fluoro-2-deoxyribose-1-phosphate and 2,2-difluoro-2-deoxyribose-1-phosphate in acidic and neutral conditions were conducted to confirm our observation. Biological evaluation of prodrugs of 2,2-difluoro-2-deoxyribose-1-phosphate for antiviral and cytotoxic activity is reported.Phosphoramidate ProTides of (fluoro) deoxyribose are reported for the first time. Their biology and stability is reported.
Co-reporter:Claire Bourdin, Christopher McGuigan, Andrea Brancale, Stanley Chamberlain, John Vernachio, Jeff Hutchins, Elena Gorovits, Alexander Kolykhalov, Jerry Muhammad, Joseph Patti, Geoffrey Henson, Blair Bleiman, K. Dawn Bryant, Babita Ganguly, Damound Hunley, Aleksandr Obikhod, C. Robin Walters, Jin Wang, Changalvala V.S. Ramamurty, Srinivas K. Battina, C. Srivinas Rao, et al.
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 7) pp:2260-2264
Publication Date(Web):1 April 2013
DOI:10.1016/j.bmcl.2012.12.004
7-Deazapurines are known to possess broad antiviral activity, however the 2′-C-methylguanosine analogue displays poor cell permeation and limited phosphorylation, thus is not an efficient inhibitor of hepatitis C virus (HCV) replication. We previously reported the 6-O-methyl entity as a prodrug moiety to increase liphophilicity of guanine nucleosides and the ProTide approach applied to 2′-C-methyl-6-O-methylguanosine has lead to potent HCV inhibitors now in clinical trials. In this Letter, we report the synthesis and biological evaluation of 2′-C-methyl-6-O-methyl-7-deaza guanosine and ProTide derivatives. In contrast to prior studies, removal of the N-7 of the nucleobase entirely negates anti-HCV activity compared to the 2′-C-methyl-6-O-methylguanosine analogues. To understand better this significant loss of activity, enzymatic assays and molecular modeling were carried out and suggested 2′-C-methyl-6-O-methyl-7-deaza guanosine and related ProTides do not act as efficient prodrugs of the free nucleotide, in marked contrast to the case of the parent guanine analogue.
Co-reporter:Michaela Serpi ; Rita Bibbo ; Stephanie Rat ; Helen Roberts ; Claire Hughes ; Bruce Caterson ; María José Alcaraz ; Anna Torrent Gibert ; Carlos Raul Alaez Verson
Journal of Medicinal Chemistry 2012 Volume 55(Issue 10) pp:4629-4639
Publication Date(Web):April 13, 2012
DOI:10.1021/jm300074y
(d)-Glucosamine and other nutritional supplements have emerged as safe alternative therapies for osteoarthritis (OA), a chronic and degenerative articular joint disease. In our preceding paper, a series of novel O-6 phosphate N-acetyl (d)-glucosamine prodrugs aimed at improving the oral bioavailability of N-acetyl-(d)-glucosamine as its putative bioactive phosphate form were shown to have greater chondroprotective activity in vitro when compared to the parent agent. In order to extend the SAR studies, this work focuses on the O-3 and O-4 phosphate prodrugs of N-acetyl-(d)-glucosamine bearing a 4-methoxy phenyl group and different amino acid esters on the phosphate moiety. Among the compounds, the (l)-proline amino acid-containing prodrugs proved to be the most active of the series, more effective than the prior O-6 compounds, and well processed in chondrocytes in vitro. Data on human cartilage support the notion that these novel O-3 and O-4 regioisomers may represent novel promising leads for drug discovery for osteoarthritis.
Co-reporter:Christopher McGuigan ; Paola Murziani ; Magdalena Slusarczyk ; Blanka Gonczy ; Johan Vande Voorde ; Sandra Liekens ;Jan Balzarini
Journal of Medicinal Chemistry 2011 Volume 54(Issue 20) pp:7247-7258
Publication Date(Web):September 5, 2011
DOI:10.1021/jm200815w
The fluorinated pyrimidine family of nucleosides continues to represent major current chemotherapeutic agents for treating solid tumors. We herein report their phosphate prodrugs, ProTides, as promising new derivatives, which partially bypass the dependence of the current drugs on active transport and nucleoside kinase-mediated activation. They are also resistant to metabolic deactivation by phosphorolytic enzymes. We report 39 ProTides of the fluorinated pyrimidine FUDR with variation in the aryl, ester, and amino acid regions. Notably, only certain ProTide motifs are successful in delivering the nucleoside monophosphate into intact cells. We also find that the ProTides retain activity in mycoplasma infected cells, unlike FUDR. Data suggest these compounds to be worthy of further progression.
Co-reporter:Christopher McGuigan ; Karolina Madela ; Mohamed Aljarah ; Claire Bourdin ; Maria Arrica ; Emma Barrett ; Sarah Jones ; Alexander Kolykhalov ; Blair Bleiman ; K. Dawn Bryant ; Babita Ganguly ; Elena Gorovits ; Geoffrey Henson ; Damound Hunley ; Jeff Hutchins ; Jerry Muhammad ; Aleksandr Obikhod ; Joseph Patti ; C. Robin Walters ; Jin Wang ; John Vernachio ; Changalvala V. S. Ramamurty ; Srinivas K. Battina ;Stanley Chamberlain
Journal of Medicinal Chemistry 2011 Volume 54(Issue 24) pp:8632-8645
Publication Date(Web):October 31, 2011
DOI:10.1021/jm2011673
We herein report phosphorodiamidates as a significant new phosphate prodrug motif. Sixty-seven phosphorodiamidates are reported of two 6-O-alkyl 2′-C-methyl guanosines, with significant variation in the diamidate structure. Both symmetrical and asymmetric phosphorodiamidates are reported, derived from various esterified amino acids, both d and l, and also from various simple amines. All of the compounds were evaluated versus hepatitis C virus in replicon assay, and nanomolar activity levels were observed. Many compounds were noncytotoxic at 100 μM, leading to high antiviral selectivities. The agents are stable in acidic, neutral, and moderately basic media and in selected biological media but show efficient processing by carboxypeptidases and efficiently yield the free nucleoside monophosphate in cells. On the basis of in vitro data, eight leads were selected for additional in vivo evaluation, with the intent of selecting one candidate for progression toward clinical studies. This phosphorodiamidate prodrug method may have broad application outside of HCV and antivirals as it offers many of the advantages of phosphoramidate ProTides but without the chirality issues present in most cases.
Co-reporter:Christopher McGuigan ; Arnaud Gilles ; Karolina Madela ; Mohamed Aljarah ; Sabrina Holl ; Sarah Jones ; John Vernachio ; Jeff Hutchins ; Brenda Ames ; K. Dawn Bryant ; Elena Gorovits ; Babita Ganguly ; Damound Hunley ; Andrea Hall ; Alexander Kolykhalov ; Yule Liu ; Jerry Muhammad ; Nicholas Raja ; Robin Walters ; Jin Wang ; Stanley Chamberlain ;Geoffrey Henson
Journal of Medicinal Chemistry 2010 Volume 53(Issue 13) pp:4949-4957
Publication Date(Web):June 8, 2010
DOI:10.1021/jm1003792
Hepatitis C virus infection constitutes a serious health problem in need of more effective therapies. Nucleoside analogues with improved exposure, efficacy, and selectivity are recognized as likely key components of future HCV therapy. 2′-C-Methylguanosine triphosphate has been known as a potent inhibitor of HCV RNA polymerase for some time, but the parent nucleoside is only moderately active due to poor intracellular phosphorylation. We herein report the application of phosphoramidate ProTide technology to bypass the rate-limiting initial phosphorylation of this nucleoside. Over 30 novel ProTides are reported, with variations in the aryl, ester, and amino acid regions. l-Alanine compounds are recognized as potent and selective inhibitors of HCV in replicon assay but lack rodent plasma stability despite considerable ester variation. Amino acid variation retaining the lead benzyl ester moiety gives an increase in rodent stability but at the cost of potency. Finally l-valine esters with ester variation lead to potent, stable compounds. Pharmacokinetic studies on these agents in the mouse reveal liver exposure to the bioactive triphosphate species following single oral dosing. Systemic exposure of the ProTide and parent nucleoside are low, indicating possible low toxicity in vivo, while liver concentrations of the active species may be predictive of efficacy in the clinic. This represents one of the most thorough cross-species studies of ProTides to date.
Co-reporter:Marco Derudas, Andrea Brancale, Lieve Naesens, Johan Neyts, Jan Balzarini, Christopher McGuigan
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 7) pp:2748-2755
Publication Date(Web):1 April 2010
DOI:10.1016/j.bmc.2010.02.015
Ribavirin is a nucleoside analogue with broad antiviral activity. Here we report the synthesis and biological evaluation of novel ribavirin ProTides designed to deliver the bioactive ribavirin monophosphate into cells. Some of the compounds display activity similar to the parent nucleoside against a range of viruses. Enzymatic, cell lysate and preliminary modeling studies have been performed to investigate the lack of enhancement of potency by the ProTides, and these indicate a failure at the final, amino acid cleavage step in the ProTide activation process, leading to inefficient release of the nucleoside monophosphate.Phosphoramidate ProTides of ribavirin do not show in vitro antiviral enhancement despite apparent efficient ester cleavage in vitro. Poor amino acid cleavage in vitro is implicated.
Co-reporter:Marco Derudas ; Davide Carta ; Andrea Brancale ; Christophe Vanpouille ; Andrea Lisco ; Leonid Margolis ; Jan Balzarini
Journal of Medicinal Chemistry 2009 Volume 52(Issue 17) pp:5520-5530
Publication Date(Web):July 31, 2009
DOI:10.1021/jm9007856
Recently, it has been reported that phosphorylated acyclovir (ACV) inhibits human immunodeficiency virus type 1 (HIV-1) reverse transcriptase in a cell-free system. To deliver phosphorylated ACV inside cells, we designed ACV monophosphorylated derivatives using ProTide technology. We found that the l-alanine derived ProTides show anti-HIV activity at noncytotoxic concentrations; ester and aryl variation was tolerated. ACV ProTides with other amino acids, other than l-phenylalanine, showed no detectable activity against HIV in cell culture. The inhibitory activity of the prodrugs against herpes simplex virus (HSV) types -1 and -2 and thymidine kinase-deficient HSV-1 revealed different structure−activity relationships but was again consistent with successful nucleoside kinase bypass. Enzymatic and molecular modeling studies have been performed in order to better understand the antiviral behavior of these compounds. ProTides showing diminished carboxypeptidase lability translated to poor anti-HIV agents and vice versa, so the assay became predictive.
Co-reporter:Christopher McGuigan, Olivier Bidet, Marco Derudas, Graciela Andrei, Robert Snoeck, Jan Balzarini
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 8) pp:3025-3027
Publication Date(Web):15 April 2009
DOI:10.1016/j.bmc.2009.03.022
Novel alkenyl substituted aryl bicyclic furano pyrimidines have been prepared and evaluated in vitro against Varicella Zoster Virus (VZV). The para-substituted analogues retain the nanomolar potency we have reported for p-alkyl analogues, while the ortho- and meta-alkenyl systems lose 3–4 orders of potency.Nanomolar potency versus VZV for para systems only. Ortho and meta poorly active.
Co-reporter:Christopher McGuigan, Plinio Perrone, Karolina Madela, Johan Neyts
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 15) pp:4316-4320
Publication Date(Web):1 August 2009
DOI:10.1016/j.bmcl.2009.05.122
β-2′-C-Methyl purines (1, 2) are known inhibitors of hepatitis C virus (HCV). We herein report the synthesis, biological and enzymatic evaluation of their 5′-phosphoramidate ProTides. Described herein are seven l-alanine phosphoramidate derivatives with variations to the amino acid ester. The 1-naphthyl phosphoramidate of β-2′-methylguanosine containing the benzyl ester (20) was the most active at 0.12 μM, an 84-fold of increase in activity compared to the parent nucleoside (2) with no increase of cytotoxicity. The carboxypeptidase mediated hydrolysis of several ProTides showed a predictive correlation with their activity versus HCV in replicon.
Co-reporter:Christopher McGuigan, Mary Rose Kelleher, Plinio Perrone, Sinead Mulready, Giovanna Luoni, Felice Daverio, Sonal Rajyaguru, Sophie Le Pogam, Isabel Najera, Joseph A. Martin, Klaus Klumpp, David B. Smith
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 15) pp:4250-4254
Publication Date(Web):1 August 2009
DOI:10.1016/j.bmcl.2009.05.099
We report the design, synthesis and evaluation of a family of ca 50 phosphoramidate ProTides of the potent anti-HCV compound 4′-azidocytidine (R1479), with variation on the ester, amino acid and aryl moiety of the ProTide. Sub-μM inhibitors of HCV emerge. The compounds are all non-cytotoxic in the replicon assay. We herein report detailed SARs for each of the regions of the ProTide.Fifty ProTides prepared. Leads have HCV EC50 ca 0.5 μM and CC50 >100 μM.
Co-reporter:Youcef Mehellou Dr.;Jan Balzarini
ChemMedChem 2009 Volume 4( Issue 11) pp:1779-1791
Publication Date(Web):
DOI:10.1002/cmdc.200900289
Abstract
Prodrug technologies aimed at delivering nucleoside monophosphates into cells (protides) have proved to be effective in improving the therapeutic potential of antiviral and anticancer nucleosides. In these cases, the nucleoside monophosphates are delivered into the cell, where they may then be further converted (phosphorylated) to their active species. Herein, we describe one of these technologies developed in our laboratories, known as the phosphoramidate protide method. In this approach, the charges of the phosphate group are fully masked to provide efficient passive cell-membrane penetration. Upon entering the cell, the masking groups are enzymatically cleaved to release the phosphorylated biomolecule. The application of this technology to various therapeutic nucleosides has resulted in improved antiviral and anticancer activities, and in some cases it has transformed inactive nucleosides to active ones. Additionally, the phosphoramidate technology has also been applied to numerous antiviral nucleoside phosphonates, and has resulted in at least three phosphoramidate-based nucleotides progressing to clinical investigations. Furthermore, the phosphoramidate technology has been recently applied to sugars (mainly glucosamine) in order to improve their therapeutic potential. The development of the phosphoramidate technology, mechanism of action and the application of the technology to various monophosphorylated nucleosides and sugars will be reviewed.
Co-reporter:Christopher McGuigan, Felice Daverio, Isabel Nájera, Joseph A. Martin, Klaus Klumpp, David B. Smith
Bioorganic & Medicinal Chemistry Letters 2009 19(11) pp: 3122-3124
Publication Date(Web):
DOI:10.1016/j.bmcl.2009.03.138
Co-reporter:Christopher McGuigan ; Michaela Serpi ; Rita Bibbo ; Helen Roberts ; Clare Hughes ; Bruce Caterson ; Ana Torrent Gibert ;Carlos Raúl Alaez Verson
Journal of Medicinal Chemistry 2008 Volume 51(Issue 18) pp:5807-5812
Publication Date(Web):August 21, 2008
DOI:10.1021/jm800594c
We report the application of the phosphoramidate ProTide approach, developed by us for antiviral nucleosides, to sugar derivatives with potential chondroprotection against osteoarthritis. In particular, N-acetylglucosamine was converted to a series of 06 arylaminoacyl phosphoramidates with ester and amino acid variation. Compounds were prepared by two routes, with or without sugar protection, and were isolated as phosphate diastereoisomers. The compounds were assayed for cellular toxicity and for inhibition of IL-1 induced glycosaminoglycan (GAG) release (i.e., proteoglycan degradation) from bovine articular cartilage in vitro explant cultures. By comparison to the N-acetyl glucosamine parent, some of the analogues show a significant enhancement in efficacy in the inhibition of inflammatory cytokine-induced proteoglycan degradation.
Co-reporter:Christopher McGuigan, Marco Derudas, Joachim J. Bugert, Graciela Andrei, Robert Snoeck, Jan Balzarini
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 15) pp:4364-4367
Publication Date(Web):1 August 2008
DOI:10.1016/j.bmcl.2008.06.069
Novel phosphoramidates of acyclovir have been prepared and evaluated in vitro against acyclovir-sensitive and -resistant herpes simplex virus (HSV) types 1 and 2 and varicella-zoster virus (VZV). Unlike the parent nucleoside these novel phosphate prodrugs retain antiviral potency versus the ACV-resistant virus strain, suggesting an efficient bypass of the viral thymidine kinase.Low or sub-μM versus HSV-1 and -2 and VZV. Retain full activity versus thymidine kinase mutants proving intracellular phosphate delivery.
Co-reporter:Marco D. Migliore ; Nicola Zonta ; Christopher McGuigan ; Geoffrey Henson ; Graciela Andrei ; Robert Snoeck ;Jan Balzarini
Journal of Medicinal Chemistry 2007 Volume 50(Issue 26) pp:6485-6492
Publication Date(Web):December 6, 2007
DOI:10.1021/jm070357e
Carbocyclic nucleoside analogues are catabolically stable since they are resistant to phosphorolytic cleavage by pyrimidine nucleoside phosphorylase enzymes. The carbocyclic analogue (C-BCNA) of the highly potent and selective anti-VZV bicyclic nucleoside analogue (BCNA) 6-pentylphenylfuro[2,3-d]pyrimidine-2′-deoxyribose was synthesized using carbocyclic 2′-deoxyuridine as starting material. C-BCNA was found to be chemically more stable than the furano lead, but it was shown to be significantly less antivirally active than its parent nucleoside analogue. It was noted to have a 10-fold lower inhibitory activity against the VZV-encoded thymidine kinase. This reduction of activity may be attributed to the different conformation of the sugar and base, as predicted by computational studies and supported by NMR studies. However, other factors besides affinity for VZV-TK must account for the greatly reduced antiviral potency.
Co-reporter:Youcef Mehellou, Christopher McGuigan, Andrea Brancale, Jan Balzarini
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 13) pp:3666-3669
Publication Date(Web):1 July 2007
DOI:10.1016/j.bmcl.2007.04.043
We report the synthesis of 2′,3′-didehydro-2′,3′-dideoxyuridine (d4U) and 2′,3′-dideoxyuridine (ddU) phosphoramidate ‘ProTide’ derivatives and their evaluation against HIV-1 and HIV-2. In addition, we conducted molecular modeling studies on both d4U and ddU monophosphates to investigate their second phosphorylation process. The findings from the modeling studies provide compelling evidence for the lack of anti-HIV activity of d4U phosphoramidates, in contrast with the corresponding ddU phosphoramidates.Phosphate pro-drug technologies (ProTides) lead to the activation of inactive nucleosides such as ddU, but not d4U, versus HIV. Reasons for the difference are explored, including the second phosphorylation step.
Co-reporter:Christopher McGuigan, Jean-Christophe Thiery, Felice Daverio, Wen G. Jiang, Gaynor Davies, Malcolm Mason
Bioorganic & Medicinal Chemistry 2005 Volume 13(Issue 9) pp:3219-3227
Publication Date(Web):2 May 2005
DOI:10.1016/j.bmc.2005.02.041
Based on our wide ranging knowledge of phosphoramidate ProTides as anti-viral agents we have tuned the lead anti-cancer agent thymectacin in the ester and amino acid regions and revealed a substantial enhancement in in vitro potency versus colon and prostate cancer cell lines. Twelve analogues have been reported, with yields of 29–78%. The compounds are fully characterised and data clearly reveal the presence of two phosphate diastereoisomers, as expected, in roughly equi-molar proportions. The compounds were evaluated in tissue culture versus three different tumour cell lines, using thymectacin as the control. It is notable that minor structural modification of the parent phenyl methoxyalaninyl structure of thymectacin leads to significant enhancements in potency. In particular, replacement of the methyl ester moiety in the lead by a benzyl ester gave a 175-fold boost in potency versus colon cancer HT115. This derivative emerges as a low micromolar inhibitor of HT115 cells and a new lead for further optimisation.
Co-reporter:Giovanna Luoni, Christopher McGuigan, Graciela Andrei, Robert Snoeck, Erik De Clercq, Jan Balzarini
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 16) pp:3791-3796
Publication Date(Web):15 August 2005
DOI:10.1016/j.bmcl.2005.05.071
Further to the discovery of bicyclic furanopyrimidine nucleoside analogues (BCNAs) as potent anti-VZV agents, a branched series of this family of compounds was synthesised. The aim was to study the impact of the geometry and steric hindrance in the side chain as well as lipophilic role of this moiety on biological activity. The results showed a detrimental effect of branching on antiviral activity, with a different magnitude depending on the position of branching in the side chain. This study again showed the importance of this moiety for biological activity, as well as the limited efficacy of the C log P value as a tool for predicting the potency of BCNAs, while suggesting an alternative rationale behind the design of future series.
Co-reporter:Annette Angell, Christopher McGuigan, Luis Garcia Sevillano, Robert Snoeck, Graciela Andrei, Erik De Clercq, Jan Balzarini
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 10) pp:2397-2399
Publication Date(Web):17 May 2004
DOI:10.1016/j.bmcl.2004.03.029
Thieno analogues of the potent and selective furo-pyrimidine anti-VZV nucleoside family bearing a p-alkylphenyl side chain have been synthesised and tested for their antiviral activity against Varicella–Zoster virus (VZV). While the alkyl chain analogues were shown to retain full antiviral activity against VZV, these new analogues did not when compared to their furo parent nucleosides.Thieno analogues of the potent and selective furo-pyrimidine anti-VZV nucleoside family bearing a p-alkylphenyl side chain have been synthesised and tested for their antiviral activity against Varicella–Zoster virus (VZV). While the alkyl chain analogues were shown to retain full anti-VZV activity, these new analogues did not when compared to their furo parent nucleosides.
Co-reporter:Christopher McGuigan, Andrea Brancale, Graciela Andrei, Robert Snoeck, Erik De Clercq, Jan Balzarini
Bioorganic & Medicinal Chemistry Letters 2003 Volume 13(Issue 24) pp:4511-4513
Publication Date(Web):15 December 2003
DOI:10.1016/j.bmcl.2003.08.028
Several novel bicyclic furanopyrimidine deoxy nucleosides have been designed, prepared and evaluated as anti-Varicella Zoster Virus agents. The compounds have long ether side chains. Uniquely amongst compounds of this family to date the present agents show dual anti- (VZV) and human cytomegalovirus (HCMV) activity. The lead compounds inhibit VZV at 10 nM and HCMV at 5 μM.Novel bicyclic nucleoside analogues bearing long alkyl side-chains were prepared and tested as inhibitors of VZV. In particular, analogues with long alkyoxy side chains are noted to be the first ever examples of this class on these bicyclic nucleoside analogues (BCNAs) to display dual anti-VZV and -HCMV action.
Co-reporter:Andrea Brancale, Christopher McGuigan, Berthe Algain, Pascal Savy, Rachid Benhida, Jean-Louis Fourrey, Graciela Andrei, Robert Snoeck, Erik De Clercq, Jan Balzarini
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 18) pp:2507-2510
Publication Date(Web):17 September 2001
DOI:10.1016/S0960-894X(01)00471-1
Thieno analogues of the potent and selective furo-pyrimidine anti-VZV nucleoside family are herein reported. The compounds retain full antiviral potency in comparison to the furo parent.Thieno analogues of the potent and selective furo-pyrimidine anti-VZV nucleoside family are herein reported. The compounds retain full antiviral potency in comparison to the furo parent.
Co-reporter:Sheila Srinivasan, Christopher McGuigan, Graciela Andrei, Robert Snoeck, Erik De Clercq, Jan Balzarini
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 3) pp:391-393
Publication Date(Web):2 February 2001
DOI:10.1016/S0960-894X(00)00672-7
Novel bicyclic nucleoside analogues bearing long alkyl side chains are prepared and tested as inhibitors of VZV. In particular, analogues with terminal unsaturation in the side chain are reported. Whilst terminal alkenyl derivatives are potent antivirals, the corresponding terminal alkynyls are poorly active.Novel bicyclic nucleoside analogues bearing long alkyl side chains are prepared and tested as inhibitors of VZV. In particular, analogues with terminal unsaturation in the side chain are reported. Whilst terminal alkenyl derivatives are potent antivirals, the corresponding terminal alkynyls are poorly active.
Co-reporter:Christopher McGuigan, Martin J. Slater, Nigel R. Parry, Alex Perry, Sarah Harris
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 7) pp:645-647
Publication Date(Web):3 April 2000
DOI:10.1016/S0960-894X(00)00061-5
We describe a synthesis of acyclovir-5′-(phenyl methoxy alaninyl) phosphate (2) from acyclovir (1). This compound was designed to act as a lipophilic, membrane-soluble prodrug of the free nucleotide. However, the biological activities of this derivative against a range of viruses indicated poor intracellular phosphate delivery, in marked contrast to the earlier successful delivery of several dideoxy anti-HIV nucleotides.
Co-reporter:Andrea Brancale, Christopher McGuigan, Graciela Andrei, Robert Snoeck, Erik De Clercq, Jan Balzarini
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 11) pp:1215-1217
Publication Date(Web):5 June 2000
DOI:10.1016/S0960-894X(00)00193-1
Preliminary SAR studies on the side chain of a new class of antiviral nucleosides have shown that terminal substitution in the side-chain, with a halogen atom, lead to potent and highly specific anti-VZV agents.
Co-reporter:Adam Siddiqui, Christopher McGuigan, Carlo Ballatore, Sheila Srinivasan, Erik De Clercq, Jan Balzarini
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 4) pp:381-384
Publication Date(Web):21 February 2000
DOI:10.1016/S0960-894X(99)00701-5
A range of polyether para-substituted phosphoramidates were synthesised and found to have substantially elevated aqueous solubilities compared to the underivatised parent prodrug. A 30-fold increase in aqueous solubility could be achieved without a substantial decrease of in vitro activity against HIV-1. Replacement of the aryl (i.e. phenolic) moiety by tyrosine led to a substantial enhancement in aqueous solubility but also to a decrease in antiviral potency. A previously unobserved trend was identified, relating increased aryl substituent steric bulk to decreased antiviral activity.
Co-reporter:Silvia Meneghesso, Evelien Vanderlinden, Annelies Stevaert, Christopher McGuigan, Jan Balzarini, Lieve Naesens
Antiviral Research (April 2012) Volume 94(Issue 1) pp:35-43
Publication Date(Web):April 2012
DOI:10.1016/j.antiviral.2012.01.007

![3-CYCLOPENTEN-1-OL, 2-[[DIMETHYL(1-METHYLETHOXY)SILYL]METHYL]-, (1S,2S)-](http://img.cochemist.com/ccimg/850300/850224-35-0.png)
![3-CYCLOPENTEN-1-OL, 2-[[DIMETHYL(1-METHYLETHOXY)SILYL]METHYL]-, (1S,2S)-](http://img.cochemist.com/ccimg/850300/850224-35-0_b.png)
![3-[(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-6-(4-pentylphenyl)furo[2,3-d]pyrimidin-2-one](http://img.cochemist.com/ccimg/319500/319425-66-6.png)
![3-[(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-6-(4-pentylphenyl)furo[2,3-d]pyrimidin-2-one](http://img.cochemist.com/ccimg/319500/319425-66-6_b.png)
![2H-1-Benzopyran-2-one, 4-methyl-7-[2-(4-methylphenyl)-2-oxoethoxy]-](http://img.cochemist.com/ccimg/195200/195137-90-7.png)
![2H-1-Benzopyran-2-one, 4-methyl-7-[2-(4-methylphenyl)-2-oxoethoxy]-](http://img.cochemist.com/ccimg/195200/195137-90-7_b.png)
![Uridine, 5'-O-[bis(4-methoxyphenyl)phenylmethyl]-2'-deoxy-5-fluoro-](http://img.cochemist.com/ccimg/104500/104495-48-9.png)
![Uridine, 5'-O-[bis(4-methoxyphenyl)phenylmethyl]-2'-deoxy-5-fluoro-](http://img.cochemist.com/ccimg/104500/104495-48-9_b.png)
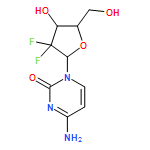
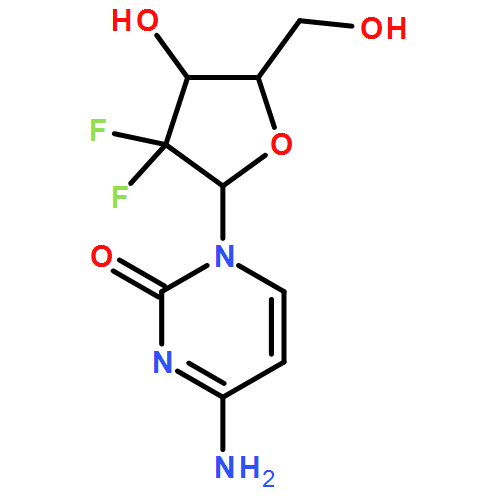
![6H-Purin-6-one,9-[[2-(acetyloxy)ethoxy]methyl]-2-amino-1,9-dihydro-](http://img.cochemist.com/ccimg/102800/102728-64-3.png)
![6H-Purin-6-one,9-[[2-(acetyloxy)ethoxy]methyl]-2-amino-1,9-dihydro-](http://img.cochemist.com/ccimg/102800/102728-64-3_b.png)
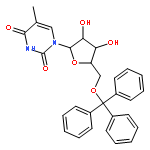
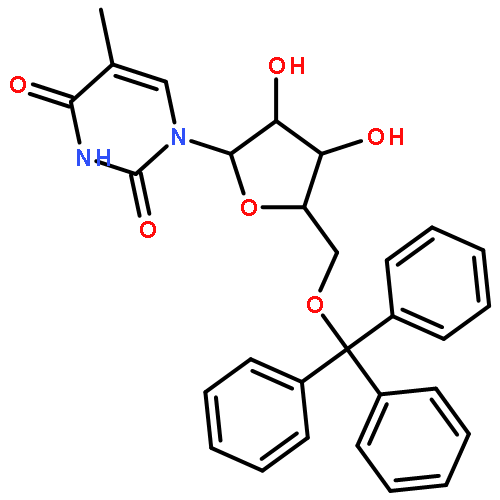
![N-(9-((6aR,8R,9R,9aS)-9-hydroxy-2,2,4,4-tetraisopropyl-tetrahydro-6H-furo[3,2-f][1,3,5,2,4]trioxadisilocin-8-yl)-9H-purin-6-yl)acetamide](http://img.cochemist.com/ccimg/85400/85335-73-5.png)
![N-(9-((6aR,8R,9R,9aS)-9-hydroxy-2,2,4,4-tetraisopropyl-tetrahydro-6H-furo[3,2-f][1,3,5,2,4]trioxadisilocin-8-yl)-9H-purin-6-yl)acetamide](http://img.cochemist.com/ccimg/85400/85335-73-5_b.png)
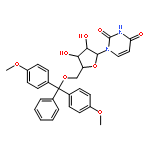
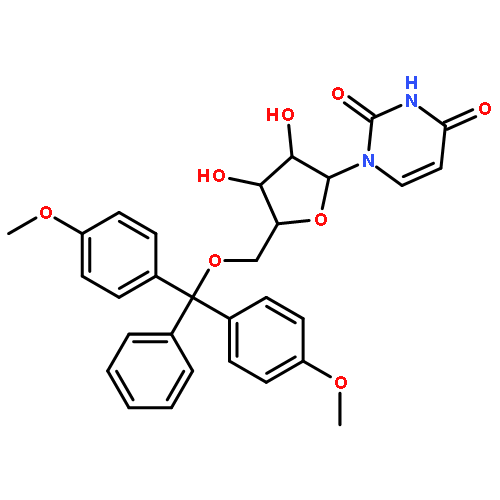
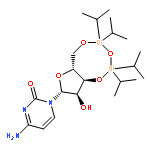
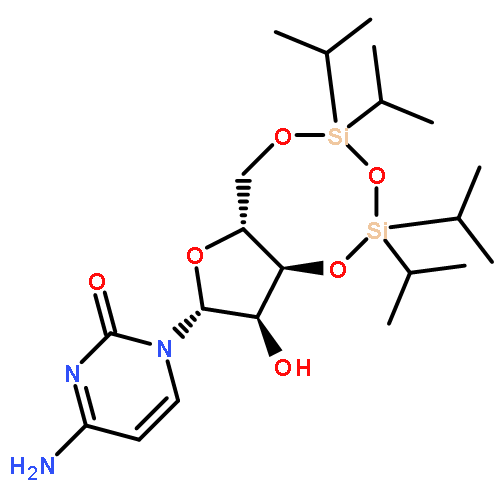







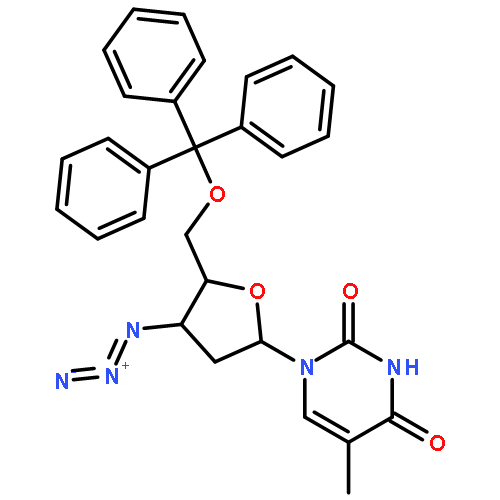



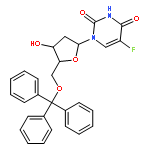
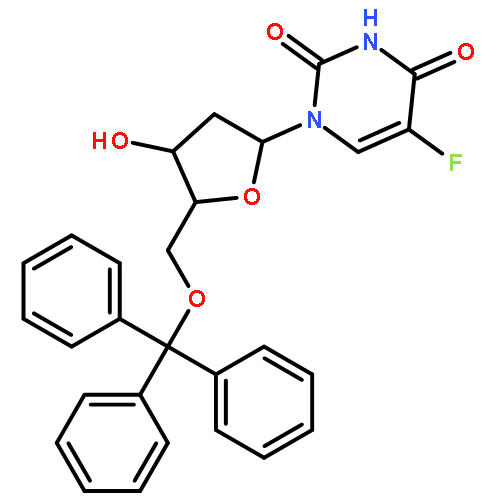
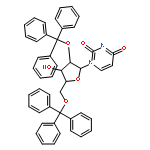
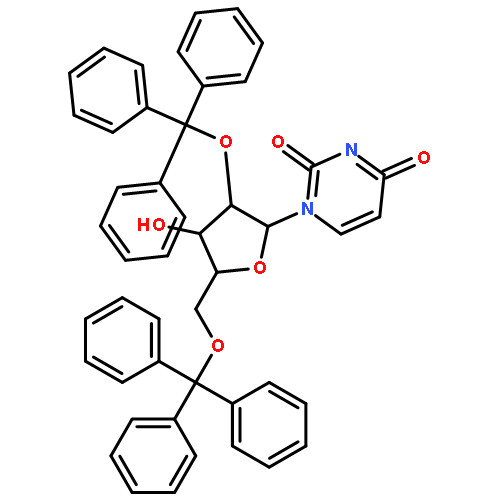


![1-[3-hydroxy-4-trityloxy-5-(trityloxymethyl)oxolan-2-yl]pyrimidine-2,4-dione](http://img.cochemist.com/ccimg/4800/4710-75-2.png)
![1-[3-hydroxy-4-trityloxy-5-(trityloxymethyl)oxolan-2-yl]pyrimidine-2,4-dione](http://img.cochemist.com/ccimg/4800/4710-75-2_b.png)






![L-ALANINE, N-[CHLORO(1-NAPHTHALENYLOXY)PHOSPHINYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/906700/906670-24-4.png)
![L-ALANINE, N-[CHLORO(1-NAPHTHALENYLOXY)PHOSPHINYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/906700/906670-24-4_b.png)