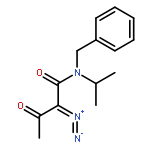
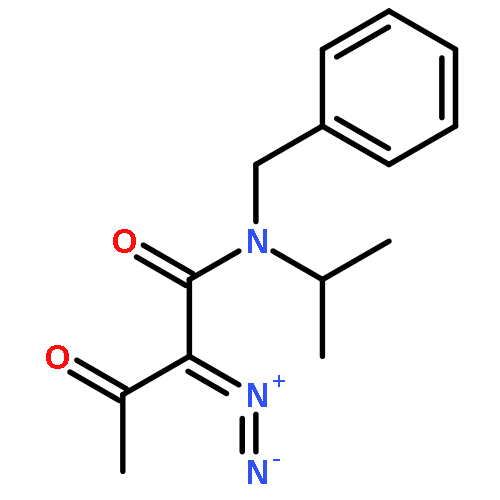
Co-reporter:Longfei Li, Ming Lei, and Shigeyoshi Sakaki
Organometallics September 25, 2017 Volume 36(Issue 18) pp:3530-3530
Publication Date(Web):September 11, 2017
DOI:10.1021/acs.organomet.7b00457
The variable coordination geometries, multiple spin states, and high density of states of first row transition metals offer a new frontier in the catalytic chemistry. A DFT study has been performed in order to unveil these characteristic features of iron metallaboratrane complex in alkene hydrogenation. A detailed spin-state analysis reveals there exist two minimum energy crossing points in the formation of (TPB)(μ-H)Fe(H) 3triplet from (TPB)Fe(N2) 1triplet. In the catalytic cycle, 3triplet at triplet state plays a role of active species, and the hydrogenation at triplet state is more favorable than that at singlet state. The dissociation of phosphine arm of TPB ligand from Fe center occurs easily in the triplet state, because the antibonding dσ is singly occupied in 3triplet. However, a nondissociative pathway without any phosphine ligand dissociation is not likely to occur. The product is formed via the σ-bond metathesis between a dihydrogen molecule and a Fe-styryl moiety. The usual direct reductive elimination involving bridging hydride (μ-H) is very difficult because the μ-H is strongly bonded with Fe and B atoms.
Co-reporter:Xuelu Ma and Ming Lei
The Journal of Organic Chemistry March 3, 2017 Volume 82(Issue 5) pp:
Publication Date(Web):February 14, 2017
DOI:10.1021/acs.joc.7b00016
The mechanism of directed hydrogenation of hydroxylated alkene catalyzed by bis(phosphine)cobalt dialkyl complexes has been studied by DFT calculations. The possible reaction channels of alkene hydrogenation catalyzed by catalytic species (0T, 0P, and 0) were investigated. The calculated results indicate that the preferred catalytic activation processes undergo a 1,2 alkene insertion. 0P and 0 prefer the β hydrogen elimination mechanism with an energy barrier of 9.5 kcal/mol, and 0T prefers the reductive elimination mechanism with an energy barrier of 11.0 kcal/mol. The second H2 coordination in the σ bond metathesis mechanism needs to break the agostic H2–βC bond of metal–alkyl intermediates (21P and 21T), which owns the larger energetic span compared to the reductive elimination. This theoretical study shows that the most favorable reaction pathway of alkene hydrogenation is the β hydrogen elimination pathway catalyzed by the planar (dppe)CoH2. The hydrogenation activity of Co(II) compounds with redox-innocent phosphine donors involves the Co(0)–Co(II) catalytic mechanism.
Co-reporter:Longfei Li, Yuhui Pan and Ming Lei
Catalysis Science & Technology 2016 vol. 6(Issue 12) pp:4450-4457
Publication Date(Web):02 Feb 2016
DOI:10.1039/C5CY01225B
A three-dimensional quantitative structure–selectivity relationship (3D-QSSR) model was developed to investigate the enantioselectivity in asymmetric ketone hydrogenation (AKH) catalyzed by RuH2(diphosphine)(diamine) complexes, through a comparative molecular field analysis (CoMFA). The predicted enantiomeric excess (ee) of the chiral alcohol products was in good agreement with the experimental ones, and the developed model showed good statistics in terms of correlation coefficients (q2 = 0.798, r2 = 0.996). The predictive power of the developed 3D-QSSR model was further proved by a test set of 5 ruthenium complexes, with an r2 of 0.974. The contour map analysis illustrated the sterically and electrostatically favored regions of the ruthenium catalysts for improving the enantioselectivity in the asymmetric hydrogenation. Under the guidance of the model, we modified the structure of the catalyst RuH2[(S)-tolbinap][(S,S)-dpen] (A1) to form the structure RuH2[(S)-tolbinap][(S,S)-dpen-NH2] (C1) where the aromatic rings of the dpen are substituted with amino groups in the para position. The theoretically predicted catalyst, C1, shows a theoretically calculated increase in the ee of AKH by 6.2%. In addition, a computational validation was performed for catalyst C1 under the density function theory (DFT), and a larger calculated difference in energy barriers in the hydrogen transfer step accounted for the enhanced enantioselectivity. In conclusion, the 3D-QSSR method could provide a plausible design criterion for the homogeneous transition-metal (TM) catalysts of asymmetric hydrogenation.
Co-reporter:Hui Li, Xuelu Ma and Ming Lei
Dalton Transactions 2016 vol. 45(Issue 20) pp:8506-8512
Publication Date(Web):11 Apr 2016
DOI:10.1039/C6DT00268D
A density functional theory (DFT) study was performed to reveal that the substituent effects in the α-site have an effect on the chemoselectivity of the intramolecular Buchner reaction of diazoacetamide catalyzed by Rh2(OAc)4. The substituent effect is investigated considering five different groups (Z = –Me, –OMe, –H, –CN and –C(O)Me) in the substrates. The substituent group in the α-site changes the electronegativity of the C-atom in carbene and affects the chemoselectivity. The basis of chemoselectivity is the distribution of products that was analyzed by DFT calculations. The barrier energy of the favorable pathway is clearly lower than that of the other pathways. Nucleophilic substituent groups, such as –H, –OMe and –Me, are regarded as electron-donating groups, which increase the electropositivity of the C-atom in carbene compounds and improve the reactivity of the aromatic addition reaction. Electrophilic substituent groups, such as –CN and –C(O)Me, are regarded as electron-withdrawing groups, which decrease the electropositivity of the C-atom in carbene compounds and favor the C–H activation step. The computational results showed that the main product is cycloheptatriene when Z = –H/–OMe. The main product is β-lactam when the substituent group is –CN/–C(O)Me. When the substituent group is –Me, the products are a mixture of γ-lactams, β-lactams and cycloheptatriene.
Co-reporter:Xinli Duan;Xin Zhang;Binglin Xu;Fang Wang
Chemical Biology & Drug Design 2016 Volume 88( Issue 1) pp:142-154
Publication Date(Web):
DOI:10.1111/cbdd.12743
Dopamine D3 receptor (D3R) is considered as a potential target for the treatment of nervous system disorders, such as Parkinson's disease. Current research interests primarily focus on the discovery and design of potent D3 agonists. In this work, we selected 40 D3R agonists as the research system. Comparative molecular field analysis (CoMFA) of three-dimensional quantitative structure–activity relationship (3D-QSAR), structure–selectivity relationship (3D-QSSR), and molecular docking was performed on D3 receptor agonists to obtain the details at atomic level. The results indicated that both the CoMFA model (r2 = 0.982, q2 = 0.503, = 0.893, SEE = 0.057, F = 166.308) for structure–activity and (r2 = 0.876, q2 = 0.436, = 0.828, F = 52.645) for structure–selectivity have good predictive capabilities. Furthermore, docking studies on three compounds binding to D3 receptor were performed to analyze the binding modes and interactions. The results elucidate that agonists formed hydrogen bond and hydrophobic interactions with key residues. Finally, we designed six molecules under the guidance of 3D-QSAR/QSSR models. The activity and selectivity of designed molecules have been improved, and ADMET properties demonstrate they have low probability of hepatotoxicity (<0.5). These results from 3D-QSAR/QSSR and docking studies have great significance for designing novel dopamine D3 selective agonists in the future.
Co-reporter:Hui Li, Xuelu Ma, Baohua Zhang, and Ming Lei
Organometallics 2016 Volume 35(Issue 19) pp:3301-3310
Publication Date(Web):September 28, 2016
DOI:10.1021/acs.organomet.6b00503
A density functional theory (DFT) study has been conducted to unveil the mechanisms of tandem oxidative acetoxylation/ortho C–H activation/carbocyclization catalyzed by Pd(OAc)2. The potential competitive reaction pathways between oxidative acetoxylation and ortho C–H activation, C–H activation with outer-sphere and inner-sphere acetate ligands, and the role of DMSO in the reaction have been discussed in detail. The calculated results indicate that the oxidative acetoxylation proceeds before ortho C–H activation in this tandem reaction in a neutral system without DMSO as a ligand coordinated to Pd. A six-membered transition state is proposed in the oxidative acetoxylation step, and a six-membered transition state is proposed in the palladium carboxylate catalyzed sp2 C–H activation step. The coordination of outer-sphere acetate ion to Pd decreases the energy barrier of the step of ortho sp2 C–H activation. In addition, this theoretical work demonstrates that the cosolvent DMSO as a ligand coordinated with Pd decreases the energy barrier of C–H activation. Also, the reaction tandem sequence changes to ortho C–H activation/oxidative acetoxylation/carbocyclization induced by DMSO as a ligand coordinated with Pd.
Co-reporter:Ming Lei;Yuhui Pan ;Xuelu Ma
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 5) pp:794-803
Publication Date(Web):
DOI:10.1002/ejic.201403027
Abstract
A density functional theory (DFT) study was performed to unveil the nature of dihydrogen (H2) production from aqueous-phase methanol dehydrogenation catalyzed by a ruthenium pincer complex. Three catalytic cycles of methanol, formaldehyde, and formate dehydrogenations were investigated at the ωB97X-D/BSI level. The calculated results indicate that the methanol-assisted hydrogen-release step is much more favorable than the direct hydrogen-release one. The dehydrogenation step, the methanol-assisted hydrogen-release step, and the CO2-release step are the rate-determining steps of methanol, formaldehyde, and formate dehydrogenations (stage I, stage II, and stage III), respectively. In addition, the formate dehydrogenation is proposed to be more difficult than the methanol and formaldehyde dehydrogenations according to calculated free-energy profiles. Methanol and formaldehyde dehydrogenation likely follow an outer-sphere mechanism, but formate dehydrogenation could involve both outer-sphere and inner-sphere mechanisms. These results add to our fundamental understanding of this efficient hydrogen-generation reaction from methanol, which could be helpful in the implementation of the methanol/hydrogen economy.
Co-reporter:Xuelu Ma, Ming Lei, and Shubin Liu
Organometallics 2015 Volume 34(Issue 7) pp:1255-1263
Publication Date(Web):March 31, 2015
DOI:10.1021/om501316t
Transition metal complexes play a key role in creating efficient molecular catalysis processes leading to the ammonia production from earth-abundant dinitrogen. It is indispensable to examine the mechanism and influence of transition metal complexes on dinitrogen hydrogenation. In this paper, the mechanism of the dinitrogen hydrogenation triggered by bimetallic complexes [L2M]2(μ-η2:η2-N2) (M = Ta and Zr, L = Sita-type or Chirik-type ligand) is investigated by density functional theory calculation. Our results show that the side-on ditantalum dinitrogen complex with Sita-type ligands favors the pathway of homolytic dihydrogen splitting when hydrogenation products are generated. However, the dihydrogen splitting switches to the heterolytic pathway as the dominant mechanism when Zr is the metal center. The ditantalum dinitrogen complex undergoes hydrogenation much easier from the side-on coordination mode than the side-on-end-on mode with Sita-type or Chirik-type ligands. With these findings from the computational study, this work identifies that different metal centers and coligands (Sita-type or Chirik-type) in different binding modes (side-on-end-on or side-on bridged) dictate the pathway of dihydrogen cleavage triggered by the bimetallic complexes. This work should provide insights on factors impacting the dinitrogen hydrogenation process and shed light on designing new transition metals and ligands for dinitrogen hydrogenation catalysts in the future.
Co-reporter:Xinli Duan, Min Zhang, Xin Zhang, Fang Wang, Ming Lei
Journal of Molecular Graphics and Modelling 2015 Volume 57() pp:143-155
Publication Date(Web):April 2015
DOI:10.1016/j.jmgm.2015.01.014
•D2-like and 5-HT2A receptors were modeled using β2-adrenergic receptor as template.•Homology model refined by molecular dynamics simulations.•Docking studies show the correlation between computed and experimental pKi is good.•Evidenced D3R model is reliable by comparing modeled D3R with crystal structure.•These models will be useful in virtual screening and design of antipsychotic drugs.Psychiatric disorders, such as schizophrenia, bipolar disorder and major depression, are paid more and more attention by human due to their upward tendency in modern society. D2-like and 5-HT2A receptors have been proposed as targets of antipsychotic drugs. Atypical antipsychotic drugs have been deemed to improve the treatment of positive, negative and extrapyramidal symptoms. Unfortunately, no experimental structures for these receptors are available except D3 receptor (D3R). Therefore, it is necessary to construct structures of D2-like and 5-HT2A receptors to investigate the interaction between these receptors and their antagonists. Accordingly, homology models of dopamine D2, D3, D4 and serotonin 5-HT2A receptors have been built on the high-resolution crystal structure of the β2-adrenergic receptor, and refined by molecular dynamics simulations. The backbone root-mean-square deviation (RMSD) of D3R model relative to crystal structure is 1.3 Å, which proves the reliability of homology modeling. Docking studies reveal that the binding modes of four homology models and their antagonists are consistent with experimental site-directed mutagenesis data. The calculated pKi values agree well with the experimental pKi ones. Antagonists with linear structures such as butyrophenones and benzisoxazolyl piperidines are easily docked into D2-like and 5-HT2A receptors. Polycyclic aromatic compounds have weaker affinity with four receptors. Homology models of D2-like and 5-HT2A receptors will be helpful for predicting the affinity of novel ligands, and could be used as three-dimensional (3D) templates for antipsychotic virtual screening and further drug discovery.D2, D3, D4 and 5-HT2A receptors were modeled and refined by molecular dynamics simulations. Docking studies show the correlations between computed and experimental pKi is matched well. The homology models can be used as 3D templates for antipsychotic drug design and virtual screening in the future.
Co-reporter:Xuelu Ma, Yanhui Tang and Ming Lei
Dalton Transactions 2014 vol. 43(Issue 30) pp:11658-11666
Publication Date(Web):15 May 2014
DOI:10.1039/C4DT00646A
This work studied the bent and planar structures of M2N2 cores of a series of dinuclear early transition-metal complexes (M = Zr, Hf, Nb, Ta, Mo and W) containing a side-on bridging dinitrogen ligand using DFT method. The calculated results propose three key factors favoring a bent structure: (1) the availability of a single electron in the metal centers which leads to the bonding interaction between two metal atoms, (2) no remarkable steric effect around the metal centers, and (3) the cis conformation of the ligands in the dinitrogen dinuclear complexes. In addition, the bent and planar structures of M2N2 could be transformed into each other if the steric hindrance was slight.
Co-reporter:Ming Lei, Zhidong Wang, Xiaojie Du, Xin Zhang, and Yanhui Tang
The Journal of Physical Chemistry A 2014 Volume 118(Issue 39) pp:8960-8970
Publication Date(Web):April 15, 2014
DOI:10.1021/jp501941b
Asymmetric hydroformylation (AHF) catalyzed by transition metal (TM) complexes bearing chiral phosphorus ligands is one of the most powerful synthetic ways that could provide chiral aldehydes directly from alkenes and syngas in one step. Experiments have proved the efficiency of Rh catalyst with hybrid phosphorus ligands owning two different phosphorus moieties in AHF. Herein the origin of enantioselectivity of AHF catalyzed by RhH(CO)2[(R,S)-Yanphos] was studied at M06/BSI level using the density functional theory (DFT) method to unveil a fundamental understanding on factors contributing to the efficiency in AHF. The alkene insertion step is supposed to be the chirality-determining step in the whole catalytic cycle of the Rh-Yanphos system. Four possible pathways of styrene (Sub1) insertion step (pathways R1, S1, R2, and S2) were discussed; the calculated results indicate that pathways R1 and S2 are proposed to be two dominant alkene insertion pathways and that styrene tends to adopt apical coordination mode (A mode) to Rh center in pathways R1 and S2 compared to equatorial coordination mode (E mode) in pathways R2 and S1. The enantioselectivity of AHFs of ten alkene substrates (CH2═CH–R, R═Ph, C(═O)OCH3, Ph-(p)-Me, Ph-(p)-OMe, Ph-(p)-iBu, Ph-(p)-F, Ph-(p)-Cl, Ph-(o)-F, OC(═O)–Ph and O–Ph, corresponding alkenes are abbreviated as Sub1 to Sub10, respectively) were also investigated. The predicted chiralities agree well with experimental results. The present work suggests that the relative stabilities of coordination modes (A/E mode) of alkene to 2 (RhH(CO)[(R,S)-Yanphos]) might be of importance in the enantioselectivity of AHF catalyzed by Rh-Yanphos.
Co-reporter:Min Wang;Xin Zhang;Zhuo Chen;YanHui Tang
Science China Chemistry 2014 Volume 57( Issue 9) pp:1264-1275
Publication Date(Web):2014 September
DOI:10.1007/s11426-014-5102-2
In this paper, we used density functional theory (DFT) computations to study the mechanisms of the hydroacylation reaction of an aldehyde with an alkene catalyzed by Wilkinson’s catalyst and an organic catalyst 2-amino-3-picoline in cationic and neutral systems. An aldehyde’s hydroacylation includes three stages: the C-H activation to form rhodium hydride (stage I), the alkene insertion into the Rh-H bond to give the Rh-alkyl complex (stage II), and the C-C bond formation (stage III). Possible pathways for the hydroacylation originated from the trans and cis isomers of the catalytic cycle. In this paper, we discussed the neutral and cationic pathways. The rate-determining step is the C-H activation step in neutral system but the reductive elimination step in the cationic system. Meanwhile, the alkyl group migration-phosphine ligand coordination pathway is more favorable than the phosphine ligand coordination-alkyl group migration pathway in the C-C formation stage. Furthermore, the calculated results imply that an electron-withdrawing group may decrease the energy barrier of the C-H activation in the benzaldehyde hydroacylation.
Co-reporter:Xuelu Ma, Xin Zhang, Wenchao Zhang and Ming Lei
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 3) pp:901-910
Publication Date(Web):19 Nov 2012
DOI:10.1039/C2CP43401F
In this paper, the reaction mechanisms of CO assisted N2 cleavage and functionalization activated by a dinuclear hafnium complex are studied using a density function theory (DFT) method. Several key intermediates (Ia, Ib, Ic and Id) with axial/equatorial NCO coordination structures are found to be of importance along reaction pathways of CO assisted N2 functionalization, which could provide a profound theoretical insight into the C–N bond formation and N–N bond cleavage. There are two different attack directions to insert the first CO molecule into the Hf–N bonds of the dinuclear hafnium complex, which lead to C–N bond formation. The calculated results imply that CO insertion into the Hf1–N3 bond (Path A1) reacts more easily than that into the Hf2–N3 bond (Path A3). But for the insertion of the second CO insertion to give 2A, there are two possibilities (Path A1 and Path A2) according to this insertion being after/before N–N bond cleavage. Two pathways (Path A1 and Path A2) are proved to be possible to form final dinitrogen functionalized products (oxamidide 2A, 2B and 2C) in this study, which explain the formation of different oxamidide isomers in CO assisted N2 functionalization activated by a dinuclear hafnium complex.
Co-reporter:Ran Feng, Ang Xiao, Xin Zhang, Yanhui Tang and Ming Lei
Dalton Transactions 2013 vol. 42(Issue 6) pp:2130-2145
Publication Date(Web):07 Nov 2012
DOI:10.1039/C2DT32210B
In this paper, the origins of enantioselectivity in asymmetric ketone hydrogenation catalyzed by RuH2(binap)(cydn) (cydn = trans-1,2-diaminocyclohexane) were discussed. Fifteen substrates involving aromatic, heteroaromatic, olefinic and dialkyl prochiral ketones were used to probe the catalytic mechanism and find an effective way to predict the chirality of the products. The calculated results demonstrate that the hydrogen transfer (HT) step from the Ru complex to the ketone substrate is the chirality-determining step in the H2-hydrogenation of ketones. The hydrogenation of aromatic-alkyl ketones can give higher enantiomeric excess (ee) values than that of dialkyl ketones. An interesting intermediate (denoted as ABS) could be formed if there is an α-hydrogen for R/R′ groups of the ketone due to the H2–Hα interaction. Two substituent groups of the ketone could rotate around the CO axis in two directions, clockwise or counter-clockwise. This rotation, with the big or conjugative substituent group away from/toward the closer binap ligand of the Ru catalyst, will form favorable/unfavorable chiral products with an Re-/Si- intermediate structure. On the contrary, if there is no such α-hydrogen in any substituent group of the ketone, ABS and another intermediate (denoted as INT) would not exist. This study indicates that the conjugative effect of the substituent groups of the ketone play an important role in differentiating the R/R′ groups of the ketone, while steric and electrostatic effects contribute to a minor extent. Furthermore, the disparity of the R and R′ groups of the ketone is of importance in the enantioselectivity and the favorable chiral alcohol is formed when the structure of the conjugative/big substituent group is away from the closer binap ligand of the RuH2(binap)(cydn) catalyst. According to the three factors of the substituent group and the fourth quadrant theory, the enantioselectivity of 91 prochiral ketones catalyzed by a series of Ru catalysts were predicted. All of the predictions are consistent with the experimental results.
Co-reporter:Xiao-Jie Du, Yan-Hui Tang, Xin Zhang, Ming Lei
Chinese Chemical Letters 2013 Volume 24(Issue 12) pp:1083-1086
Publication Date(Web):December 2013
DOI:10.1016/j.cclet.2013.07.004
This paper studied the mechanism of the alkene insertion elementary step in the asymmetric hydroformylation (AHF) catalyzed by RhH(CO)2[(R,S)-Yanphos] using four alkene substrates (CH2CHPh, CH2CHPh(p)Me, CH2CHC(O)OCH3 and CH2CHOC(O)Ph, abbreviated as A1–A4). Interestingly, the equatorial vertical coordination mode (A mode) with respect to the Rh center was found for A1 and A2 but not for A3 and A4, although the equatorial in-plane coordination mode (E mode) was found for A1–A4. The relative energy of the E mode of the η2-intermediates is lower than that of the A mode. In the alkene insertion step, Path 1 is more favorable than Path 2 for this system. As for A1 and A2, there could be a transformation between 2eq and 2ax.This paper studied the mechanism of the alkene insertion elementary step in the asymmetric hydroformylation (AHF) catalyzed by RhH(CO)2[(R,S)-Yanphos] using four alkene substrates (CH2CHPh, CH2CHPh(p)Me, CH2CHC(O)OCH3 and CH2CHOC(O)Ph, abbreviated as A1–A4). Interestingly, the equatorial vertical coordination mode (A mode) with respect to the Rh center was found for A1 and A2 but not for A3 and A4, although the equatorial in-plane coordination mode (E mode) was found for A1–A4. The relative energy of the E mode of the η2-intermediates is lower than that of the A mode. In the alkene insertion step, Path 1 is more favorable than Path 2 for this system. As for A1 and A2, there could be a transformation between 2eq and 2ax.
Co-reporter:LiJun Zhao;LiangRen Zhang
Science China Chemistry 2013 Volume 56( Issue 11) pp:1550-1563
Publication Date(Web):2013 November
DOI:10.1007/s11426-013-4894-9
Transthyretin (TTR), a plasma protein with a tetramer structure, could form amyloid fibril associated with several human diseases through the dissociation of tetramer and the misfolding of monomer. These amyloidogenesis can be inhibited by small molecules which bind to the central channel of TTR. A number of small molecules like 2-arylbenzoxazoles (ABZ) analogues are proposed as promising therapeutic strategy to treat amyloidosis. In this work, comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) three-dimensional quantitative structure-activity relationship (3D-QSAR) and docking studies were performed on series of 2-arylbenzoxazoles (ABZ) and linker-Y analogues to investigate the inhibitory activities of TTR amyloidogenesis at atomic level. Significant correlation coefficients for ABZ series (CoMFA, r2 = 0.877, q2 = 0.431; CoMSIA, r2 = 0.836, q2 = 0.447) and those for linker-Y series (CoMFA, r2 = 0.828, q2 = 0.522; CoMSIA, r2 = 0.800, q2 = 0.493) were obtained, and the generated models were validated using test sets. In addition, docking studies on 6 compounds binding to TTR were performed to analyze the forward or reverse binding mode and interactions between molecules and TTR. These results from 3D-QSAR and docking studies have great significance for designing novel TTR amyloidogenesis inhibitors in the future.
Co-reporter:Xuelu Ma, Yanhui Tang, and Ming Lei
Organometallics 2013 Volume 32(Issue 23) pp:7077-7082
Publication Date(Web):November 19, 2013
DOI:10.1021/om4007856
A DFT study on the carboxylation of hafnocene and ansa-zirconocene dinitrogen complexes with CO2 indicates that the most favorable initial CO2 insertion into M–N (M = Hf, Zr) proceeds by a stepwise path rather than a concerted [2 + 2] path. The calculated results explain the regioselectivity of the N–C formation in experiments. In addition, a comparative analysis of ring tension and charge distribution unveils the different activities of N–N bond cleavage in the CO and CO2 direct N–C bond formation reactions.
Co-reporter:Xin Zhang, Xiaojia Guo, Yue Chen, Yanhui Tang, Ming Lei and Weihai Fang
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 17) pp:6003-6012
Publication Date(Web):06 Mar 2012
DOI:10.1039/C2CP23936A
In this paper, the mechanism of ketone hydrogenation catalyzed by five Ru bifunctional catalysts with different structural frameworks was studied in detail using density functional theory (DFT). This mechanism contains hydrogen transfer, dehydrogenation of alcohol, and dihydrogen activation fundamental reactions. The involvement of alcohol is also discussed and found with different activities in hydrogen transfer, dehydrogenation and dihydrogen activation steps in five systems. Our calculated results indicate that the weak Ru–H bond, stronger basicity of hydride and stronger X–H acidity will decrease the barrier of the HT step, and that the polar micro-environment of dihydrogen coordinating with Ru catalysts and short hydrogen transfer distance would be able to facilitate the heterolytic splitting of dihydrogen in the dihydrogen activation step.
Co-reporter:Tianhu Yuan;Xin Zhang;Zehan Hu;Fang Wang
Biopolymers 2012 Volume 97( Issue 12) pp:998-1009
Publication Date(Web):
DOI:10.1002/bip.22116
Abstract
Piscidin 1 (Pis-1) has a high broad-spectrum activity against bacteria, fungi, and viruses but it also has a moderate hemolytic activities. To improve the antibacterial activity and to reduce toxicity, mutants Pis-1AA (G8A/G13A double mutant) and Pis-1PG (G8P mutant) have been designed based on the crystal structure of Pis-1. Eighteen independent molecular dynamics (MD) simulations of Pis-1 and its mutants with membranes are conducted in this article. Furthermore, 60 independent MD simulations of three peptides in water box have also been discussed for comparison. The results indicate that the unfolding process starts at the middle of the peptide. Pis-1 disrupts easily in the region of Val10-Lys14. Pis-1PG has a flexible N-terminal region, and the interaction between N-terminal and C-terminal is very weak. Pis-1AA has the most stable helical structure. In addition, percentage of native contacts and hydrogen bonds analysis are also performed. Lipid-peptide interaction analysis suggests that Pis-1 and Pis-1AA has a stronger interaction with the zwitterionic dioleoylphosphatidylcholine (DOPC) lipid bilayer than Pis-1PG. When compared with the results of peptide with membrane, peptides are unstable and unfolding quickly in water solution. Our results are applicable in examining diversities on hemolytic, antibacterial, and selectivity of antimicrobial peptides. © 2012 Wiley Periodicals, Inc. Biopolymers 97:998–1009, 2012.
Co-reporter:Wenchao Zhang, Yanhui Tang, Ming Lei, Keiji Morokuma, and Djamaladdin G. Musaev
Inorganic Chemistry 2011 Volume 50(Issue 19) pp:9481-9490
Publication Date(Web):August 30, 2011
DOI:10.1021/ic201159z
To elucidate (i) the physicochemical properties of the {(η5-C5Me5)[TaIV](i-Pr)C(Me)N(i-Pr)}2(μ-η1:η1-N2), I, [TaIV]2(μ-η1:η1-N2), and {(η5-C5Me5)[TaV](i-Pr)C(Me)N(i-Pr)}2(μ-N)2, II, [TaV]2(μ-N)2, complexes; (ii) the mechanism of the I → II isomerization; and (iii) the reaction mechanism of these complexes with an H2 molecule, we launched density functional (B3LYP) studies of model systems 1, 2, and 3 where the C5Me5 and (i-Pr)C(Me)N(i-Pr) ligands of I (or II) were replaced by C5H5 and HC(NCH3)2, respectively. These calculations show that the lower-lying electronic states of 1, [TaIV]2(μ-η1:η1-N2), are nearly degenerate open-shell singlet and triplet states with two unpaired electrons located on the Ta centers. This finding is in reasonable agreement with experiments [J. Am Chem. Soc.2007, 129, 9284–9285] showing easy accessibility of paramagnetic and diamagnetic states of I. The ground electronic state of the bis(μ-nitrido) complex 2, [TaV]2(μ-N)2, is a closed-shell singlet state in agreement with the experimentally reported diamagnetic feature of II. The 1-to-2 rearrangement is a multistep and highly exothermic process. It occurs with a maximum of 28.7 kcal/mol free energy barrier required for the (μ-η1:η1-N2) → (μ-η2:η2-N2) transformation step. Reaction of 1 with H2 leading to the 1,4-addition product 3 proceeds with a maximum of 24.2 kcal/mol free energy barrier associated by the (μ-η1:η1-N2) → (μ-η2:η1-N2) isomerization step. The overall reaction 1 + H2 → 3 is exothermic by 20.0 kcal/mol. Thus, the addition of H2 to 1 is kinetically and thermodynamically feasible and proceeds via the rate-determining (μ-η1:η1-N2) → (μ-η2:η1-N2) isomerization step. The bis(μ-nitrido) complex 2, [TaV]2(μ-N)2, does not react with H2 because of the large energy barrier (49.5 kcal/mol) and high endothermicity of the reaction. This conclusion is also in excellent agreement with the experimental observation [J. Am Chem. Soc.2007, 129, 9284–9285].
Co-reporter:Xiaojia Guo, Yanhui Tang, Xin Zhang, and Ming Lei
The Journal of Physical Chemistry A 2011 Volume 115(Issue 44) pp:12321-12330
Publication Date(Web):October 5, 2011
DOI:10.1021/jp2046728
In this paper, the mechanism of transfer hydrogenation of acetophenone catalyzed by ruthenium–acetamido complex was studied using density function theory (DFT) method. The catalytic cycle of transfer hydrogenation consists of hydrogen transfer (HT) step and dehydrogenation (DH) step of isopropanol (IPA). Inner sphere mechanism (paths 1 and 7) and outer sphere mechanism (paths 2–6) in HT step are fully investigated. Calculated results indicate that DH step of IPA (from i1 to i2) is the rate-determining step in the whole catalytic cycle, which has a potential energy barrier of 16.2 kcal/mol. On the other hand, the maximum potential energy barriers of paths 1–7 in the HT step are 5.9, 12.7, 24.4, 16.8, 23.7, 7.2, and 6.1 kcal/mol, respectively. The inner sphere pathways (paths 1 and 7) are favorable hydrogen transfer modes compared with outer sphere pathways, and the proton transferred to the oxygen atom of acetophenone comes from the hydroxyl group but not from amino group of acetamido ligand. Those theoretical results are in agreement with experimental report. However, in view of this DFT study in the inner sphere mechanism of HT step, hydride transfer and proton transfer are concerted and asynchronous hydrogen transfer but not a stepwise one, and hydride transfer precedes proton transfer in this case.
Co-reporter:Ming Lei, Wenchao Zhang, Yue Chen and Yanhui Tang
Organometallics 2010 Volume 29(Issue 3) pp:543-548
Publication Date(Web):January 8, 2010
DOI:10.1021/om900434n
In this work, H2 activation processes in hydrogenation of ketones catalyzed by late transition metal−ligand bifunctional catalysts have been studied using the DFT method. For systems A (RuH2-diphosephine/diamine complex) and B (Ru-η5-Cp*-1,2-diamine complex), the dihydrogen activation process in neutral and basic conditions (path 1) consisted of two steps: H2 coordination and H−H cleavage. However, dihydrogen activations catalyzed by complexes C−F (Ru-η6-arene and Rh/Ir-cyclopentadiene complexes) along path 1 consist of only H−H cleavage due to the absence of H2 coordination. Thus, systems C−F have higher energy barriers (ΔG >27 kcal/mol) for dihydrogen activation than systems A and B. However, for systems C−F under acidic conditions, dihydrogen activation (path 2) consists of the two steps involving H2 coordination; thus the dihydrogen activation barriers decrease greatly, resulting in an easy splitting of H2. These results agree well with experiments. In the conversion from transfer hydrogenation to H2 hydrogenation for C−F, the protonation of 16e complex MNC−F changes the N2−M1−Y3 (Y = N or O) delocalized π-bond into a M1−Y3 localized π-bond. Therefore, the 16e complexes, which can provide a vacant site for H2 coordination, tend to perform H2 hydrogenation.
Co-reporter:Yue Chen, Yanhui Tang and Ming Lei
Dalton Transactions 2009 (Issue 13) pp:2359-2364
Publication Date(Web):03 Feb 2009
DOI:10.1039/B815699A
Three modes for hydride and proton transfer were observed in the hydrogen transfer step in ketone hydrogenation catalyzed by transition metal-centered diphosphine–diamine complexes. The results indicate that a weaker metal–hydride bond would lead to hydride transfer preceding proton transfer with a low energy barrier, and that a strong MN bond in transition metal amido complexes could increase the energy barrier for the dihydrogen activation step.
Co-reporter:Yue Chen, Yanhui Tang, Shubin Liu, Ming Lei and Weihai Fang
Organometallics 2009 Volume 28(Issue 7) pp:2078-2084
Publication Date(Web):March 18, 2009
DOI:10.1021/om8012212
Density functional theory calculations have been performed to study the hydrogenation of ketones catalyzed by a η6-arene ruthenium(II) complex in an acidic conditions. Six possible dihydrogen activation (DA) pathways were investigated in this work. The direct DA (path 1) and alcohol-assisted DA (path 2), which will occur in basic/neutral conditions, have higher energy barriers, 19.9 and 18.7 kcal/mol, respectively. If an acid participates in DA in the other three paths (paths 4, 5, and 6), the barrier will substantially decrease to about 3.7, 7.1, and 7.8 kcal/mol, respectively. Compared with paths 1 and 2, the acid-assisted pathways are more favorable. In path 1 and path 2, molecular hydrogen is unable to coordinate with Ru to form a stable η2-H2 ruthenium(II) complex, leading to the increase of the free energy barrier of DA. On the contrary, dihydrogen forms a stable η2-H2 ruthenium(II) complex with Ru in paths 4, 5, and 6. These results indicate that the coordination of dihydrogen with Ru plays an important role in the conversion of H2-hydrogenation in acidic conditions.
Co-reporter:Baohua Zhang, Tianhu Yuan, Hua Jiang and Ming Lei
The Journal of Physical Chemistry B 2009 Volume 113(Issue 31) pp:10934-10941
Publication Date(Web):July 14, 2009
DOI:10.1021/jp9033358
The polymer of 7-amino-8-fluoro-2-quinoline carboxylic acid (abbreviated as QFn, n denotes polymerization degree, where n = 4 or 8) can form artificial helices that do not exist in nature. The crystal structures of the quadruplex of QF4 and the duplex of QF8 (abbreviated as QP_4 and DP_8, respectively) have been unveiled by some researchers recently. In this paper, QP_4 and DP_8 were built based on their crystal structures. Though the quadruplex of QF8 and the duplex of QF4 (abbreviated as QP_8 and DP_4, respectively) were not observed in experiments, they were built using molecular modeling tools. The molecular dynamics (MD) method was used to study the stabilities and assembly mechanisms of these helices at different temperatures. In MD simulations, the disassembled QP_4 was observed above 480 K. The results indicate that these oligoamide helices showed slippage of strands to some extent and that double-helical oligoamides act as a basic block in the slippage during the formation of the quadruple-helical structure. DP_4 and QP_4 showed dynamical features with helix-handedness inversion and following slippage of strands at higher temperatures. In addition, the π−π conjugations between the different chains are supposed to be responsible for the stability of the helices.
Co-reporter:Yue Chen, Shubin Liu and Ming Lei
The Journal of Physical Chemistry C 2008 Volume 112(Issue 35) pp:13524-13527
Publication Date(Web):2017-2-22
DOI:10.1021/jp8003807
Three different reaction pathways—concerted hydride transfer, concerted proton transfer, and stepwise hydrogen transfer—in the carbonyl hydrogenation step of the catalytic cycle for ketone hydrogenation catalyzed by Ru complexes are investigated in this work. Our results from this study suggest that hydride and proton asynchronous transfers in the ketone hydrogenation process are stepwise in general. It is the nature of the substrate, electron withdrawing effect, and characteristics of catalytic ligands that make one of two transition states disappear in the concerted mechanism.
Co-reporter:Ying Liu, Fang Wang, Tianwei Tan, Ming Lei
Analytica Chimica Acta 2007 Volume 581(Issue 1) pp:137-146
Publication Date(Web):2 January 2007
DOI:10.1016/j.aca.2006.08.015
In this paper, a simplified model was set up to give an insight into the properties of molecularly imprinted polymer (MIP) at molecular level using MMFF94 force field. Based on our model, the interaction energies (ΔEs) between monomers and template or its analogues were calculated, and the most possible conformations of template or its analogues interacting with monomers in the molar ratio 1/4 were found. The obtained results using the computational and conformational analysis showed that large ΔE meant more activity sites in the cavities in the resultant polymer giving high affinity and good selectivity, leading to a large imprinting factor and when the ΔE differences were small, the imprinting factors were mainly determined by the activity sites. These were well consistent with the experimental results, which confirmed the validity of the model and method proposed that were believed to benefit screening molecularly imprinted systems rapidly in an experiment-free way instead of trial-and-error approach. Considering the affinity and selectivity, 2,6-bisacrylamide pyridine was predicted to be the optimal monomer used to prepare paracetamol MIP for application in quantification of drugs from the ΔE and possible activity sites.
Co-reporter:Hao Cao, Li Deng, Ming Lei, Fang Wang, Tianwei Tan
Journal of Molecular Catalysis B: Enzymatic (November 2014) Volume 109() pp:101-108
Publication Date(Web):1 November 2014
DOI:10.1016/j.molcatb.2014.08.013
•Transient open conformation of YLLIP2 was obtained by MD simulations.•Effects of temperature and solvent microenvironment on catalytic activity of YLLIP2 were unveiled at atomic level.•Excessive temperature tended to disturb the interaction between His289 and Asp230.•Solvent tended to disturb the interaction between His289 and Ser162.•Results provided an optimized model for choosing better reaction conditions.The influence of temperature and solvent on the activity of Yarrowia lipolytica Lipase 2 (YLLIP2) was investigated. This was done by interpreting experimental results with theoretical molecular modeling of the enzyme structure by using molecular dynamic (MD) simulation. The transient open conformation of YLLIP2 was obtained. It was employed for exploring the structural rearrangement of the lid and the catalytic triad (Ser162, Asp230, and His289) at different temperatures and in different solvents. The calculated results indicated that the opened extent of the lid was positively correlated with temperature and the structural rearrangement of the catalytic triad was the crucial factor for the decreased activity of YLLIP2 at higher temperature. The polar solvent molecule approaches the catalytic triad of YLLIP2 more easily and has a stronger interaction with His289 than the non-polar solvent molecule. The interaction between His289 and Asp230 was affected by the higher temperature (333 K) whereas the interaction between His289 and Ser162 was affected by the polar solvent molecule (acetone and ethanol).Download full-size image
Co-reporter:Zehan Hu, Yanhui Tang, Houfang Wang, Xu Zhang, Ming Lei
Archives of Biochemistry and Biophysics (15 July 2008) Volume 475(Issue 2) pp:140-147
Publication Date(Web):15 July 2008
DOI:10.1016/j.abb.2008.04.024
Co-reporter:Houfang Wang, Yanhui Tang, Ming Lei
Archives of Biochemistry and Biophysics (1 October 2007) Volume 466(Issue 1) pp:85-97
Publication Date(Web):1 October 2007
DOI:10.1016/j.abb.2007.07.010
Co-reporter:Yue Chen, Yanhui Tang and Ming Lei
Dalton Transactions 2009(Issue 13) pp:NaN2364-2364
Publication Date(Web):2009/02/03
DOI:10.1039/B815699A
Three modes for hydride and proton transfer were observed in the hydrogen transfer step in ketone hydrogenation catalyzed by transition metal-centered diphosphine–diamine complexes. The results indicate that a weaker metal–hydride bond would lead to hydride transfer preceding proton transfer with a low energy barrier, and that a strong MN bond in transition metal amido complexes could increase the energy barrier for the dihydrogen activation step.
Co-reporter:Ran Feng, Ang Xiao, Xin Zhang, Yanhui Tang and Ming Lei
Dalton Transactions 2013 - vol. 42(Issue 6) pp:NaN2145-2145
Publication Date(Web):2012/11/07
DOI:10.1039/C2DT32210B
In this paper, the origins of enantioselectivity in asymmetric ketone hydrogenation catalyzed by RuH2(binap)(cydn) (cydn = trans-1,2-diaminocyclohexane) were discussed. Fifteen substrates involving aromatic, heteroaromatic, olefinic and dialkyl prochiral ketones were used to probe the catalytic mechanism and find an effective way to predict the chirality of the products. The calculated results demonstrate that the hydrogen transfer (HT) step from the Ru complex to the ketone substrate is the chirality-determining step in the H2-hydrogenation of ketones. The hydrogenation of aromatic-alkyl ketones can give higher enantiomeric excess (ee) values than that of dialkyl ketones. An interesting intermediate (denoted as ABS) could be formed if there is an α-hydrogen for R/R′ groups of the ketone due to the H2–Hα interaction. Two substituent groups of the ketone could rotate around the CO axis in two directions, clockwise or counter-clockwise. This rotation, with the big or conjugative substituent group away from/toward the closer binap ligand of the Ru catalyst, will form favorable/unfavorable chiral products with an Re-/Si- intermediate structure. On the contrary, if there is no such α-hydrogen in any substituent group of the ketone, ABS and another intermediate (denoted as INT) would not exist. This study indicates that the conjugative effect of the substituent groups of the ketone play an important role in differentiating the R/R′ groups of the ketone, while steric and electrostatic effects contribute to a minor extent. Furthermore, the disparity of the R and R′ groups of the ketone is of importance in the enantioselectivity and the favorable chiral alcohol is formed when the structure of the conjugative/big substituent group is away from the closer binap ligand of the RuH2(binap)(cydn) catalyst. According to the three factors of the substituent group and the fourth quadrant theory, the enantioselectivity of 91 prochiral ketones catalyzed by a series of Ru catalysts were predicted. All of the predictions are consistent with the experimental results.
Co-reporter:Xuelu Ma, Yanhui Tang and Ming Lei
Dalton Transactions 2014 - vol. 43(Issue 30) pp:NaN11666-11666
Publication Date(Web):2014/05/15
DOI:10.1039/C4DT00646A
This work studied the bent and planar structures of M2N2 cores of a series of dinuclear early transition-metal complexes (M = Zr, Hf, Nb, Ta, Mo and W) containing a side-on bridging dinitrogen ligand using DFT method. The calculated results propose three key factors favoring a bent structure: (1) the availability of a single electron in the metal centers which leads to the bonding interaction between two metal atoms, (2) no remarkable steric effect around the metal centers, and (3) the cis conformation of the ligands in the dinitrogen dinuclear complexes. In addition, the bent and planar structures of M2N2 could be transformed into each other if the steric hindrance was slight.
Co-reporter:Hui Li, Xuelu Ma and Ming Lei
Dalton Transactions 2016 - vol. 45(Issue 20) pp:NaN8512-8512
Publication Date(Web):2016/04/11
DOI:10.1039/C6DT00268D
A density functional theory (DFT) study was performed to reveal that the substituent effects in the α-site have an effect on the chemoselectivity of the intramolecular Buchner reaction of diazoacetamide catalyzed by Rh2(OAc)4. The substituent effect is investigated considering five different groups (Z = –Me, –OMe, –H, –CN and –C(O)Me) in the substrates. The substituent group in the α-site changes the electronegativity of the C-atom in carbene and affects the chemoselectivity. The basis of chemoselectivity is the distribution of products that was analyzed by DFT calculations. The barrier energy of the favorable pathway is clearly lower than that of the other pathways. Nucleophilic substituent groups, such as –H, –OMe and –Me, are regarded as electron-donating groups, which increase the electropositivity of the C-atom in carbene compounds and improve the reactivity of the aromatic addition reaction. Electrophilic substituent groups, such as –CN and –C(O)Me, are regarded as electron-withdrawing groups, which decrease the electropositivity of the C-atom in carbene compounds and favor the C–H activation step. The computational results showed that the main product is cycloheptatriene when Z = –H/–OMe. The main product is β-lactam when the substituent group is –CN/–C(O)Me. When the substituent group is –Me, the products are a mixture of γ-lactams, β-lactams and cycloheptatriene.
Co-reporter:Longfei Li, Yuhui Pan and Ming Lei
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 12) pp:NaN4457-4457
Publication Date(Web):2016/02/02
DOI:10.1039/C5CY01225B
A three-dimensional quantitative structure–selectivity relationship (3D-QSSR) model was developed to investigate the enantioselectivity in asymmetric ketone hydrogenation (AKH) catalyzed by RuH2(diphosphine)(diamine) complexes, through a comparative molecular field analysis (CoMFA). The predicted enantiomeric excess (ee) of the chiral alcohol products was in good agreement with the experimental ones, and the developed model showed good statistics in terms of correlation coefficients (q2 = 0.798, r2 = 0.996). The predictive power of the developed 3D-QSSR model was further proved by a test set of 5 ruthenium complexes, with an r2 of 0.974. The contour map analysis illustrated the sterically and electrostatically favored regions of the ruthenium catalysts for improving the enantioselectivity in the asymmetric hydrogenation. Under the guidance of the model, we modified the structure of the catalyst RuH2[(S)-tolbinap][(S,S)-dpen] (A1) to form the structure RuH2[(S)-tolbinap][(S,S)-dpen-NH2] (C1) where the aromatic rings of the dpen are substituted with amino groups in the para position. The theoretically predicted catalyst, C1, shows a theoretically calculated increase in the ee of AKH by 6.2%. In addition, a computational validation was performed for catalyst C1 under the density function theory (DFT), and a larger calculated difference in energy barriers in the hydrogen transfer step accounted for the enhanced enantioselectivity. In conclusion, the 3D-QSSR method could provide a plausible design criterion for the homogeneous transition-metal (TM) catalysts of asymmetric hydrogenation.
Co-reporter:Zhuo Chen, Yue Chen, Yanhui Tang and Ming Lei
Dalton Transactions 2010 - vol. 39(Issue 8) pp:NaN2043-2043
Publication Date(Web):2010/01/13
DOI:10.1039/B917934H
In this paper, the catalytic activities of RuHX(diamine)(PPh3)2 complexes with different X ligands (X = NCMe, CO, Cl, OMe, OPh, CCMe and H, corresponding catalytic processes are abbreviated in A, B, C, D, E, F and G systems, respectively) in the H2-hydrogenation of ketones were investigated using density functional theory (DFT) method. Calculated results indicate that the rate-determining step in the whole catalytic cycle is hydrogen transfer (HT) for A–E but dihydrogen activation (DA) for F and G. The free energy barriers of the HT step for A–G are 36.1, 32.3, 21.2, 14.9, 21.9, 9.4 and 6.9 kcal mol−1, respectively. The DA step consists of hydrogen coordination (HC) and hydrogen splitting (HS) steps if dihydrogen coordinates with the Ru center. The transition states (TSs) of H2 coordinating with the Ru atom for A–G except B are located. The free energy barriers of DA for A–G are 17.8 (17.8, 2.6), 21.5, 12.8 (12.8, 3.8), 12.2 (11.2, 6.2), 13.6 (13.6, 4.1), 17.1 (9.7, 7.5) and 22.0 (10.4, 11.0) kcal mol−1, respectively (the data in parentheses correspond to the barriers of HC and HS). HT barriers correlate well with the charges of hydride (H) in complex 1. HC barriers are closely related to the RuN1 double bond in 4, and HS are in line with the proton-moved-distances (PMDs) from 5 to TS5-1. This study demonstrates that catalysts D, F and G show better catalytic activities than the others, which is in good agreement with experimental results.
Co-reporter:Xin Zhang, Xiaojia Guo, Yue Chen, Yanhui Tang, Ming Lei and Weihai Fang
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 17) pp:NaN6012-6012
Publication Date(Web):2012/03/06
DOI:10.1039/C2CP23936A
In this paper, the mechanism of ketone hydrogenation catalyzed by five Ru bifunctional catalysts with different structural frameworks was studied in detail using density functional theory (DFT). This mechanism contains hydrogen transfer, dehydrogenation of alcohol, and dihydrogen activation fundamental reactions. The involvement of alcohol is also discussed and found with different activities in hydrogen transfer, dehydrogenation and dihydrogen activation steps in five systems. Our calculated results indicate that the weak Ru–H bond, stronger basicity of hydride and stronger X–H acidity will decrease the barrier of the HT step, and that the polar micro-environment of dihydrogen coordinating with Ru catalysts and short hydrogen transfer distance would be able to facilitate the heterolytic splitting of dihydrogen in the dihydrogen activation step.
Co-reporter:Xuelu Ma, Xin Zhang, Wenchao Zhang and Ming Lei
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 3) pp:NaN910-910
Publication Date(Web):2012/11/19
DOI:10.1039/C2CP43401F
In this paper, the reaction mechanisms of CO assisted N2 cleavage and functionalization activated by a dinuclear hafnium complex are studied using a density function theory (DFT) method. Several key intermediates (Ia, Ib, Ic and Id) with axial/equatorial NCO coordination structures are found to be of importance along reaction pathways of CO assisted N2 functionalization, which could provide a profound theoretical insight into the C–N bond formation and N–N bond cleavage. There are two different attack directions to insert the first CO molecule into the Hf–N bonds of the dinuclear hafnium complex, which lead to C–N bond formation. The calculated results imply that CO insertion into the Hf1–N3 bond (Path A1) reacts more easily than that into the Hf2–N3 bond (Path A3). But for the insertion of the second CO insertion to give 2A, there are two possibilities (Path A1 and Path A2) according to this insertion being after/before N–N bond cleavage. Two pathways (Path A1 and Path A2) are proved to be possible to form final dinitrogen functionalized products (oxamidide 2A, 2B and 2C) in this study, which explain the formation of different oxamidide isomers in CO assisted N2 functionalization activated by a dinuclear hafnium complex.

![Benzamide,3-chloro-5-ethyl-N-[[(2S)-1-ethyl-2-pyrrolidinyl]methyl]-6-hydroxy-2-methoxy-](http://img.cochemist.com/ccimg/84300/84226-12-0.png)
![Benzamide,3-chloro-5-ethyl-N-[[(2S)-1-ethyl-2-pyrrolidinyl]methyl]-6-hydroxy-2-methoxy-](http://img.cochemist.com/ccimg/84300/84226-12-0_b.png)