Co-reporter:Amanda L. Blasius;Roberto Baccala;Keiko Ozato;Dwight H. Kono;Rosana Gonzalez-Quintial;Ivo Rimann;Bruce Beutler
PNAS 2013 Volume 110 (Issue 8 ) pp:2940-2945
Publication Date(Web):2013-02-19
DOI:10.1073/pnas.1222798110
In vitro evidence suggests that plasmacytoid dendritic cells (pDCs) are intimately involved in the pathogenesis of lupus.
However, it remains to be determined whether these cells are required in vivo for disease development, and whether their contribution
is restricted to hyperproduction of type I IFNs. To address these issues, we created lupus-predisposed mice lacking the IFN
regulatory factor 8 (IRF8) or carrying a mutation that impairs the peptide/histidine transporter solute carrier family 15,
member 4 (SLC15A4). IRF8-deficient NZB mice, lacking pDCs, showed almost complete absence of anti-nuclear, anti-chromatin,
and anti-erythrocyte autoantibodies, along with reduced kidney disease. These effects were observed despite normal B-cell
responses to Toll-like receptor (TLR) 7 and TLR9 stimuli and intact humoral responses to conventional T-dependent and -independent
antigens. Moreover, Slc15a4 mutant C57BL/6-Faslpr mice, in which pDCs are present but unable to produce type I IFNs in response to endosomal TLR ligands, also showed an absence
of autoantibodies, reduced lymphadenopathy and splenomegaly, and extended survival. Taken together, our results demonstrate
that pDCs and the production of type I IFNs by these cells are critical contributors to the pathogenesis of lupus-like autoimmunity
in these models. Thus, IRF8 and SLC15A4 may provide important targets for therapeutic intervention in human lupus.
Co-reporter:Argyrios N. Theofilopoulos,
Rosana Gonzalez-Quintial,
Brian R. Lawson,
Yi T. Koh,
Michael E. Stern,
Dwight H. Kono,
Bruce Beutler
&
Roberto Baccala
Nature Reviews Rheumatology 2010 6(3) pp:146
Publication Date(Web):2010-02-09
DOI:10.1038/nrrheum.2009.278
Evidence strongly suggests that excessive or protracted signaling, or both, by cell-surface or intracellular innate immune receptors is central to the pathogenesis of most autoimmune and autoinflammatory rheumatic diseases. The initiation of aberrant innate and adaptive immune responses in autoimmune diseases can be triggered by microbes and, at times, by endogenous molecules—particularly nucleic acids and related immune complexes—under sterile conditions. By contrast, most autoinflammatory syndromes are generally dependent on germline or de novo gene mutations that cause or facilitate inflammasome assembly. The consequent production of proinflammatory cytokines, principally interferon-α/β and tumor necrosis factor in autoimmune diseases, and interleukin-1β in autoinflammatory diseases, leads to the creation of autoamplification feedback loops and chronicity of these syndromes. These findings have resulted in a critical reappraisal of pathogenetic mechanisms, and provide a basis for the development of novel diagnostic and therapeutic modalities for these diseases.
Co-reporter:Roberto Baccala,
Rosana Gonzalez-Quintial,
Brian R. Lawson,
Michael E. Stern,
Dwight H. Kono,
Bruce Beutler
&
Argyrios N. Theofilopoulos
Nature Reviews Rheumatology 2009 5(8) pp:448
Publication Date(Web):2009-07-14
DOI:10.1038/nrrheum.2009.136
The discovery of molecular sensors that enable eukaryotes to recognize microbial pathogens and their products has been a key advance in our understanding of innate immunity. A tripartite sensing apparatus has developed to detect danger signals from infectious agents and damaged tissues, resulting in an immediate but short-lived defense response. This apparatus includes Toll-like receptors, retinoid acid-inducible gene-I-like receptors and other cytosolic nucleic acid sensors, and nucleotide-binding and oligomerization domain-like receptors; adaptors, kinases and other signaling molecules are required to elicit effective responses. Although this sensing is beneficial to the host, excessive activation and/or engagement by self molecules might induce autoimmune and other inflammatory disorders.
Co-reporter:Brian R. Lawson, Rosana Gonzalez-Quintial, Theodoros Eleftheriadis, Michael A. Farrar, Stephen D. Miller, Karsten Sauer, Dorian B. McGavern, Dwight H. Kono, Roberto Baccala, Argyrios N. Theofilopoulos
Clinical Immunology (December 2015) Volume 161(Issue 2) pp:260-269
Publication Date(Web):1 December 2015
DOI:10.1016/j.clim.2015.08.007
•Optimal activation CD4+ T cells both in vitro and in vivo was found to require IL-7.•IL-7 signaling pathways intersect and synergize with the TCR to promote activation.•IL-7Rα blockade preferentially induced apoptosis in recently engaged CD4+ T cells.•Anti-IL-7Rα inhibited induction, progression and relapses of EAE, a model of MS.•Findings should be widely applicable to CD4+ T cell-mediated responses and diseases.IL-7 is known to be vital for T cell homeostasis but has previously been presumed to be dispensable for TCR-induced activation. Here, we show that IL-7 is critical for the initial activation of CD4+ T cells in that it provides some of the necessary early signaling components, such as activated STAT5 and Akt. Accordingly, short-term in vivo IL-7Rα blockade inhibited the activation and expansion of autoantigen-specific CD4+ T cells and, when used to treat experimental autoimmune encephalomyelitis (EAE), prevented and ameliorated disease. Our studies demonstrate that IL-7 signaling is a prerequisite for optimal CD4+ T cell activation and that IL-7R antagonism may be effective in treating CD4+ T cell-mediated neuroinflammation and other autoimmune inflammatory conditions.
.jpg)
![Decanoic acid,1,1'-[(1aR,1bS,4aS,7aS,7bS,8R,9R,9aS)-1,1a,1b,4,4a,5,7a,7b,8,9-decahydro-4a,7b-dihydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-5-oxo-9aH-cyclopropa[3,4]benz[1,2-e]azulene-9,9a-diyl]ester](http://img.cochemist.com/ccimg/27600/27536-56-7.png)
![Decanoic acid,1,1'-[(1aR,1bS,4aS,7aS,7bS,8R,9R,9aS)-1,1a,1b,4,4a,5,7a,7b,8,9-decahydro-4a,7b-dihydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-5-oxo-9aH-cyclopropa[3,4]benz[1,2-e]azulene-9,9a-diyl]ester](http://img.cochemist.com/ccimg/27600/27536-56-7_b.png)
![5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,9,9a-bis(benzoyloxy)-1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b-dihydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-,(1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-](http://img.cochemist.com/ccimg/25500/25405-85-0.png)
![5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,9,9a-bis(benzoyloxy)-1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b-dihydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-,(1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-](http://img.cochemist.com/ccimg/25500/25405-85-0_b.png)
![Decanoic acid,(1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-1,1a,1b,4,4a,5,7a,7b,8,9-decahydro-4a,7b-dihydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-5-oxo-9aH-cyclopropa[3,4]benz[1,2-e]azulene-9,9a-diylester](http://img.cochemist.com/ccimg/25000/24928-17-4.png)
![Decanoic acid,(1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-1,1a,1b,4,4a,5,7a,7b,8,9-decahydro-4a,7b-dihydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-5-oxo-9aH-cyclopropa[3,4]benz[1,2-e]azulene-9,9a-diylester](http://img.cochemist.com/ccimg/25000/24928-17-4_b.png)


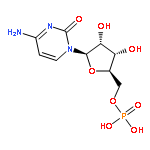
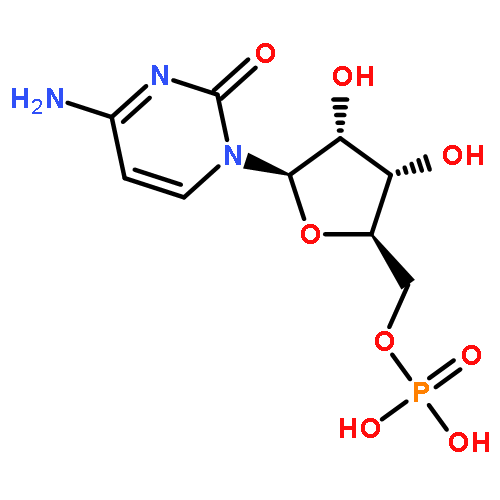


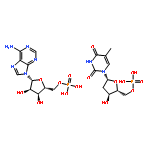
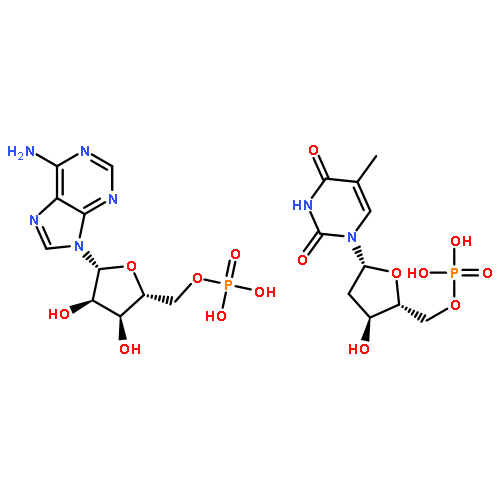
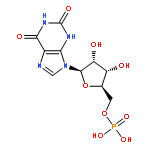
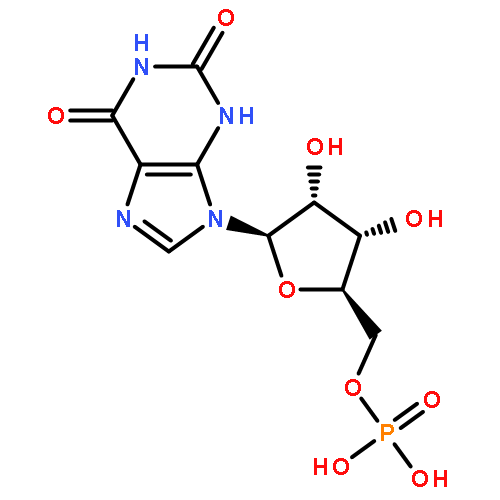


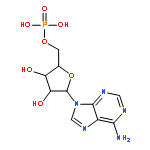

![5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b,9,9a-tetrahydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-,(1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-](http://img.cochemist.com/ccimg/17700/17673-25-5.png)
![5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-4a,7b,9,9a-tetrahydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-,(1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-](http://img.cochemist.com/ccimg/17700/17673-25-5_b.png)








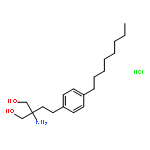




![8-Azabicyclo[3.2.1]octane,3-(diphenylmethoxy)-8-methyl-, (3-endo)-](http://img.cochemist.com/ccimg/100/86-13-5.png)
![8-Azabicyclo[3.2.1]octane,3-(diphenylmethoxy)-8-methyl-, (3-endo)-](http://img.cochemist.com/ccimg/100/86-13-5_b.png)

