Co-reporter:Fan-kun Meng
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 1) pp:197-206
Publication Date(Web):2016/12/20
DOI:10.1039/C6OB02195F
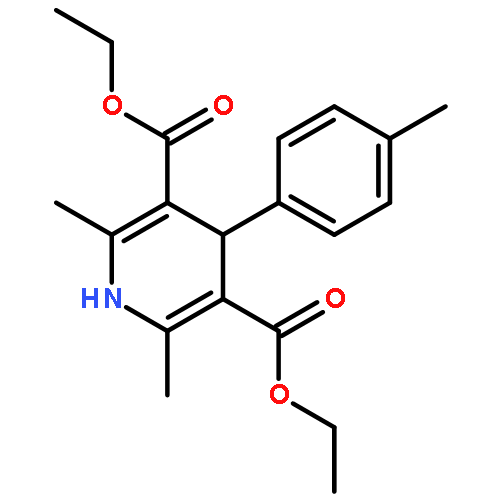
25 Dihydropyrimidine derivatives, a new class of organo-hydrides, were designed and synthesized by the Biginelli reaction. For the first time, the thermodynamic driving forces of the six elemental steps to obtain a hydride in acetonitrile were determined by isothermal titration and electrochemical methods, respectively. The effects of molecular structures and substituents on these thermodynamic parameters were examined, uncovering some interesting structure–reactivity relationships. Both the thermodynamic and kinetic studies show that the hydride transfer from dihydropyrimidines to 9-phenylxanthylium (PhXn+ClO4−) prefers a concerted mechanism.
Co-reporter:Guang-Bin Shen, Ke Xia, Xiu-Tao Li, Jun-Ling Li, Yan-Hua Fu, Lin Yuan, and Xiao-Qing Zhu
The Journal of Physical Chemistry A 2016 Volume 120(Issue 11) pp:1779-1799
Publication Date(Web):March 3, 2016
DOI:10.1021/acs.jpca.5b10135
In this work, kinetic isotope effect (KIEself) values of 68 hydride self-exchange reactions, XH(D) + X+ → X+ + XH(D), in acetonitrile at 298 K were determined using a new experimental method. KIE values of 4556 hydride cross transfer reactions, XH(D) + Y+ → X+ + YH(D), in acetonitrile were estimated from the 68 determined KIEself values of hydride self-exchange reactions using a new KIE relation formula derived from Zhu’s kinetic equation and the reliability of the estimations was verified using different experimental methods. A new KIE kinetic model to explain and predict KIE values was developed according to Zhu’s kinetic model using two different Morse free energy curves instead of one Morse free energy curve in the traditional KIE theories to describe the free energy changes of X–H bond and X–D bond dissociation in chemical reactions. The most significant contribution of this paper to KIE theory is to build a new KIE kinetic model, which can be used to not only uniformly explain the various (normal, enormous and inverse) KIE values but also safely prodict KIE values of various chemical reactions.
Co-reporter:Nan-Ping Lei, Yan-Hua Fu and Xiao-Qing Zhu
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 47) pp:11472-11485
Publication Date(Web):29 Sep 2015
DOI:10.1039/C5OB01715G
A series of analogues of indazolium alkaloids were designed and synthesized. The thermodynamic driving forces of the 6 elemental steps for the analogues of indazolium alkaloids to obtain hydride in acetonitrile were determined using an isothermal titration calorimeter (ITC) and electrochemical methods, respectively. The effects of molecular structure and substituents on the thermodynamic driving forces of the 6 steps were examined. Meanwhile, the oxidation mechanism of NADH coenzyme by indazolium alkaloids was examined using the chemical mimic method. The result shows that the oxidation of NADH coenzyme by indazolium alkaloids in vivo takes place by one-step concerted hydride transfer mechanism.
Co-reporter:Ke Xia, Guang-Bin Shen and Xiao-Qing Zhu
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 22) pp:6255-6268
Publication Date(Web):28 Apr 2015
DOI:10.1039/C5OB00538H
32 F420 coenzyme models with alkylation of the three different N atoms (N1, N3 and N10) in the core structure (XFH−) were designed and synthesized and the thermodynamic driving forces (defined in terms of the molar enthalpy changes or the standard redox potentials in this work) of the 32 XFH− releasing hydride ions, hydrogen atoms and electrons, the thermodynamic driving forces of the 32 XFH˙ releasing protons and hydrogen atoms and the thermodynamic driving forces of XF−˙ releasing electrons in acetonitrile were determined using titration calorimetry and electrochemical methods. The effects of the methyl group at N1, N3 and N10 and a negative charge on N1 and N10 atoms on the six thermodynamic driving forces of the F420 coenzyme models and their related reaction intermediates were examined; the results show that seating arrangements of the methyl group and the negative charge have remarkably different effects on the thermodynamic properties of the F420 coenzyme models and their related reaction intermediates. The effects of the substituents at C7 and C8 on the six thermodynamic driving forces of the F420 coenzyme models and their related reaction intermediates were also examined; the results show that the substituents at C7 and C8 have good Hammett linear free energy relationships with the six thermodynamic parameters. Meanwhile, a reasonable determination of possible reactions between members of the F420 family and NADH family in vivo was given according to a thermodynamic analysis platform constructed using the elementary step thermodynamic parameter of F420 coenzyme model 2FH− and NADH model MNAH releasing hydride ions in acetonitrile. The information disclosed in this work can not only fill a gap in the chemical thermodynamics of F420 coenzyme models as a class of very important organic sources of electrons, hydride ions, hydrogen atoms and protons, but also strongly promote the fast development of the chemistry and applications of F420 coenzyme.
Co-reporter:San-Min Si
The Journal of Physical Chemistry C 2015 Volume 119(Issue 1) pp:62-74
Publication Date(Web):December 4, 2014
DOI:10.1021/jp5089756
In this work, five substituted 1-phenyl-2-benzensulfonyl ethynes and the corresponding five substituted 1-phenyl-2-benzensulfonyl ethenes were designed and synthesized as representatives of the polar alkynes and the polar alkenes. Thermodynamic driving forces of eight elementary steps for reductions of the substituted ethynes and ethenes to the corresponding alkenes and alkanes in acetonitrile were determined. The differences of chemical properties between the alkynes and the alkenes as well as their various derived reaction intermediates were quantitatively examined or compared according to the determined thermodynamic driving forces of the eight elementary steps. The relative C–C π-bond heterolytic and homolytic dissociation energies of the alkynes and alkenes in acetonitrile were estimated according to the difference of the hydride affinities and hydrogen atom affinities of the related chemical species. The relative effective charges on the active center atom of the alkynes and the alkenes as well as their derived various reaction intermediates, which can be used to quantitatively measure the polarity of the corresponding chemical species, were estimated according to the Hammett substituent effects using the Hammett-type linear free energy relationships. Molecule ID Cards of the alkynes and the alkenes in acetonitrile were constructed from the determined thermodynamic driving forces of the eight elementary steps. The thermodynamic tendencies and detailed mechanisms for the reductions of the alkynes and alkenes by Hantzsch ester in acetonitrile were diagnosed according to the thermodynamic analytic platforms that were made of the Molecule ID Cards of the related reactants. It is clear that the results of this work are not only to provide good guidance for synthetic chemists to safely choose a suitable reducing agent for selective reductions of alkynes and alkenes and to rationally examine the reaction mechanisms but also to facilitate theoretical chemists to develop novel calculation methods to examine the chemistry of alkynes and alkenes.
Co-reporter:Xiao-Qing Zhu, Fei-Huang Deng, Jin-Dong Yang, Xiu-Tao Li, Qiang Chen, Nan-Ping Lei, Fan-Kun Meng, Xiao-Peng Zhao, Su-Hui Han, Er-Jun Hao and Yuan-Yuan Mu
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 36) pp:6071-6089
Publication Date(Web):16 Jul 2013
DOI:10.1039/C3OB40831K
A classical but new kinetic equation to estimate activation energies of various hydride transfer reactions was developed according to transition state theory using the Morse-type free energy curves of hydride donors to release a hydride anion and hydride acceptors to capture a hydride anion and by which the activation energies of 187 typical hydride self-exchange reactions and more than thirty thousand hydride cross transfer reactions in acetonitrile were safely estimated in this work. Since the development of the kinetic equation is only on the basis of the related chemical bond changes of the hydride transfer reactants, the kinetic equation should be also suitable for proton transfer reactions, hydrogen atom transfer reactions and all the other chemical reactions involved with breaking and formation of chemical bonds. One of the most important contributions of this work is to have achieved the perfect unity of the kinetic equation and thermodynamic equation for hydride transfer reactions.
Co-reporter:Xiao-Qing Zhu;Jin-Dong Yang
Journal of Physical Organic Chemistry 2013 Volume 26( Issue 3) pp:271-273
Publication Date(Web):
DOI:10.1002/poc.3078
Marcus theory is a well-known kinetic theory developed by Rudolph A. Marcus in 1956 for outer sphere electron transfer reactions according to the solvent polarization, which has been extensively used to describe the kinetics of various chemical reactions. However, when Marcus theory was examined with scientific principles, it is found that Marcus theory directly conflicts with the law of conservation of energy. Copyright © 2013 John Wiley & Sons, Ltd.
Co-reporter:Ying Cao, Song-Chen Zhang, Min Zhang, Guang-Bin Shen, and Xiao-Qing Zhu
The Journal of Organic Chemistry 2013 Volume 78(Issue 14) pp:7154-7168
Publication Date(Web):June 21, 2013
DOI:10.1021/jo4010926
A series of 69 polar olefins with various typical structures (X) were synthesized and the thermodynamic affinities (defined in terms of the molar enthalpy changes or the standard redox potentials in this work) of the polar olefins obtaining hydride anions, hydrogen atoms, and electrons, the thermodynamic affinities of the radical anions of the polar olefins (X•–) obtaining protons and hydrogen atoms, and the thermodynamic affinities of the hydrogen adducts of the polar olefins (XH•) obtaining electrons in acetonitrile were determined using titration calorimetry and electrochemical methods. The pure C═C π-bond heterolytic and homolytic dissociation energies of the polar olefins (X) in acetonitrile and the pure C═C π-bond homolytic dissociation energies of the radical anions of the polar olefins (X•–) in acetonitrile were estimated. The remote substituent effects on the six thermodynamic affinities of the polar olefins and their related reaction intermediates were examined using the Hammett linear free-energy relationships; the results show that the Hammett linear free-energy relationships all hold in the six chemical and electrochemical processes. The information disclosed in this work could not only supply a gap of the chemical thermodynamics of olefins as one class of very important organic unsaturated compounds but also strongly promote the fast development of the chemistry and applications of olefins.
Co-reporter:Xiao-Qing Zhu, Xiu-Tao Li, Su-Hui Han, and Lian-Rui Mei
The Journal of Organic Chemistry 2012 Volume 77(Issue 10) pp:4774-4783
Publication Date(Web):April 23, 2012
DOI:10.1021/jo3005952
The effects of substituents on the temperature dependences of kinetic isotope effect (KIE) for the reactions of the hydride transfer from the substituted 5-methyl-6-phenyl-5,6-dihydrophenanthridine (G-PDH) to thioxanthylium (TX+) in acetonitrile were examined, and the results show that the temperature dependences of KIE for the hydride transfer reactions can be converted by adjusting the nature of the substituents in the molecule of the hydride donor. In general, electron-withdrawing groups can make the KIE to have normal temperature dependence, but electron-donating groups can make the KIE to have abnormal temperature dependence. Thermodynamic analysis on the possible pathways of the hydride transfer from G-PDH to TX+ in acetonitrile suggests that the transfers of the hydride anion in the reactions are all carried out by the concerted one-step mechanism whether the substituent is an electron-withdrawing group or an electron-donating group. But the examination of Hammett-type free energy analysis on the hydride transfer reactions supports that the concerted one-step hydride transfer is not due to an elementary chemical reaction. The experimental values of KIE at different temperatures for the hydride transfer reactions were modeled by using a kinetic equation formed according to a multistage mechanism of the hydride transfer including a returnable charge-transfer complex as the reaction intermediate; the real mechanism of the hydride transfer and the root that why the temperature dependences of KIE can be converted as the nature of the substituents are changed were discovered.
Co-reporter:Xiaozhen Han, Weifang Hao, Xiao-Qing Zhu, and Vernon D. Parker
The Journal of Organic Chemistry 2012 Volume 77(Issue 15) pp:6520-6529
Publication Date(Web):July 3, 2012
DOI:10.1021/jo301042d
One representative type of heterocyclic compound that can release a hydride ion is 7,8-dihydro-9-methylcaffeine (CAFH). The one-electron oxidation potential of CAFH [−0.294 (V vs Fc+/0)] and the one-electron reduction potential of CAF+ [−2.120 (V vs Fc+/0)] were obtained using two different methods, CV and OSWV. Applying titration calorimetry data in thermodynamic cycles, the enthalpies of CAFH releasing a hydride ion [57.6 kcal/mol] and releasing a hydrogen atom [80.3 kcal/mol] and of its radical cation CAFH•+ releasing a proton [33.0 kcal/mol] and releasing a hydrogen atom [38.4 kcal/mol] have been determined. Several conclusions can be drawn from the thermodynamic results: (1) CAFH is a very good single-electron donor whose single-electron oxidation potential is much less positive than that of NAD(P)H model compound BNAH [Eox = 0.219 V vs Fc+/0]. (2) The single-electron reduction potential of CAF+ is much more negative than that of BNA+ [Ered = −1.419 V], which means that CAF+ is not a good electron acceptor. Furthermore, CAFH is a very good hydride donor compared to BNAH. The results of non-steady-state kinetic studies, for the reaction of CAFH and AcrH+ClO4–, show that the ratio of t0.50/t0.05 is larger than 13.5 and the ratio of kinit/kpfo is larger than 1. The pseudo-first-order rate constants obtained at different reaction stages decrease with the time, and the kinetic isotope was observed to be small at a short reaction time and slowly increases to 3.72 with the progress of the reaction. These kinetic results clearly display that the hydride transfer of CAFH to AcrH+ in acetonitrile is not a one-step mechanism, while the thermodynamic results show that CAFH is a very good electron donor. The combination of the kinetic results with the thermodynamics analysis shows that the hydride transfer of the caffeine derivative CAFH takes place by a two-step reversible mechanism and there is an intermediate in the reaction.
Co-reporter:ChaoTun Cao;Yue Tan
Science China Chemistry 2012 Volume 55( Issue 10) pp:2054-2056
Publication Date(Web):2012 October
DOI:10.1007/s11426-012-4679-6
Heterolytic and homolytic C-D bond dissociation energies of three NADH models: BNAH-4,4-d2, HEH-4,4-d2 and Acrd2 in acetonitrile were first estimated by using an efficient method. The results showed that the heterolytic C-D bond dissociation energies are 65.2, 70.2, and 81.9 kcal/mol and the homolytic C-D bond dissociation energies are 72.66, 70.69, and 74.95 kcal/mol for BNAH-4,4-d2, HEH-4,4-d2, and AcrD2, respectively. According to the bond dissociation energy differences of isotope isomers, an interesting conclusion can be made that the primary kinetic isotope effects are dependent not only on the zero-point energy difference of the isotope isomers, but also on the types of C-D bond dissociations, and the C-D bond homolytic dissociations should have much larger primary kinetic isotope effects (26.9–28.8) than the corresponding C-D bond heterolytic dissociations (3.9–5.4).
Co-reporter:Xiao-Qing Zhu, Jian Zhou, Chun-Hua Wang, Xiu-Tao Li, and Sha Jing
The Journal of Physical Chemistry B 2011 Volume 115(Issue 13) pp:3588-3603
Publication Date(Web):March 15, 2011
DOI:10.1021/jp200095g
5,7-Ditert-butyl-3-(3,4-dimethylphenyl)benzofuran-2(3H)-one (HP-136) (1H) and its 30 analogues (2H−5H) as benzofuranone-typical antioxidants were synthesized. The structures of the benzofuranones in solid and solution were examined by using experimental and theoretical methods. The results show that the dominant structure is the lactone form rather than the enol form both in solid and solution. The thermodynamic driving forces of the 31 benzofuranone-typical compounds to release protons [ΔGPD(XH)], hydrogen atoms [ΔGHD(XH)], and electrons [Eox(XH)] and the thermodynamic driving forces of the anions (X−) of the benzofuranones to release electrons [Eox(X−)] were determined for the first time in DMSO. The ΔGHD(XH) scale of these compounds in DMSO ranges from 65.2 to 74.1 (kcal/mol) for 1H−4H and from 73.8 to 75.0 (kcal/mol) for 5H, respectively, which are all smaller than that of the most widely used commercial antioxidant BHT (2,6-ditert-butyl-4-methylphenol, 81.6 kcal/mol), suggesting that the 31 XH could be used as good hydrogen-atom-donating antioxidants. The ΔGPD(XH) were observed to range from 11.5 to 16.0 (kcal/mol) for 1H−4H and from 18.6 to 22.4 (kcal/mol) for 5H, indicating that benzofuranones (1H−4H) are good proton donors, and their analogues (5H) should belong to middle-strong proton donors. Eox(XH) of the 31 XH to release an electron vary from 1.346 to 1.962 (V versus Fc+/0), implying that the 31 XH are weak electron donors, whereas the quite negative Eox(X−) show that X− are good electron donors. The Gibbs free-energy changes of the radical cations (XH+•) to release protons [ΔGPD(XH+•)] were evaluated according to the corresponding thermodynamic cycle, and the results reveal that XH+• are good proton donors. Further inspection of our experimental results showed the ΔGHD(XH), ΔGPD(XH), ΔGPD(XH+•), Eox(XH), and Eox(X−) of the five chemical and electrochemical processes are all linearly dependent on the sum of Hammett substituent parameters σ with very good correlation coefficients, indicating that for any one- or multisubstituted species at the para- and/or meta-position of benzofuranones and their various reaction intermediates, the five thermodynamic driving force parameters all can be easily and safely estimated from the corresponding Hammett substituent parameters. The rates of hydrogen atom transfer from XH to DPPH• were determined by using the UV−vis absorption spectroscopy technique. Combining these important thermodynamic parameters and dynamic determination results, the mechanism of hydrogen transfer from HP-136 and its analogues to DPPH• was studied. The results suggest that the hydrogen transfer from HP-136 and its analogues 2H to DPPH• actually includes two steps, proton transfer and the following electron transfer, but the proton transfer is rate-determined.
Co-reporter:Xiao-Qing Zhu, Yuan-Yuan Mu, and Xiu-Tao Li
The Journal of Physical Chemistry B 2011 Volume 115(Issue 49) pp:14794-14811
Publication Date(Web):October 30, 2011
DOI:10.1021/jp2067974
Ascorbic acid (AscH2) and dihydronicotinamide adenine dinucleotide (NADH) are two very important natural redox cofactors, which can be used as hydride, electron, and hydrogen atom sources to take part in many important bioreduction processes in vivo. The differences of the two natural reducing agents as hydride, hydrogen atom, and electron donors in thermodynamics, kinetics, and mechanisms were examined by using 5,6-isopropylidene ascorbate (iAscH–) and β-d-glucopyranosyl-1,4-dihydronicotinamide acetate (GluNAH) as their models, respectively. The results show that the hydride-donating ability of iAscH– is smaller than that of GluNAH by 6.0 kcal/mol, but the electron-donating ability and hydrogen-donating ability of iAscH– are larger than those of GluNAH by 20.8 and 8.4 kcal/mol, respectively, which indicates that iAscH– is a good electron donor and a good hydrogen atom donor, but GluNAH is a good hydride donor. The kinetic intrinsic barrier energy of iAscH– to release hydride anion in acetonitrile is larger than that of GluNAH to release hydride anion in acetonitrile by 6.9 kcal/mol. The mechanisms of hydride transfer from iAscH– and GluNAH to phenylxanthium perchlorate (PhXn+), a well-known hydride acceptor, were examined, and the results show that hydride transfer from GluNAH adopted a one-step mechanism, but the hydride transfer from iAscH– adopted a two-step mechanism (e–H•). The thermodynamic relation charts (TRC) of the iAscH– family (including iAscH–, iAscH•, iAsc•–, and iAsc) and of the GluNAH family (including GluNAH, GluNAH•+, GluNA•, and GluNA+) in acetonitrile were constructed as Molecule ID Cards of iAscH– and of GluNAH in acetonitrile. By using the Molecule ID Cards of iAscH– and GluNAH, the character chemical properties not only of iAscH– and GluNAH but also of the various reaction intermediates of iAscH– and GluNAH all have been quantitatively diagnosed and compared. It is clear that these comparisons of the thermodynamics, kinetics, and mechanisms between iAscH– and GluNAH as hydride and electron donors in acetonitrile should be quite important and valuable to diagnose and understand the different roles and functions of ascorbic acid and NADH as hydride, hydrogen atom, and electron sources in vivo.
Co-reporter:Xiao-Qing Zhu, Yue Tan and Chao-Tun Cao
The Journal of Physical Chemistry B 2010 Volume 114(Issue 5) pp:2058-2075
Publication Date(Web):January 14, 2010
DOI:10.1021/jp911137p
A series of 45 dihydropyridine-type organic compounds as hydride source were designed and synthesized. The thermodynamic driving forces (defined as enthalpy changes or redox potentials in this work) of the dihydropyridines to release hydride anions, hydrogen atoms (hydrogen for short), and electrons in acetonitrile, the thermodynamic driving forces of the radical cations of the dihydropyridines to release protons and hydrogens in acetonitrile, and the thermodynamic driving forces of the neutral pyridine-type radicals of the dihydropyridines to release electron in acetonitrile were determined by using titration calorimetry and electrochemical methods. The rates and activation parameters of hydride transfer from the dihydropyridines to acridinium perclorate, a well-known hydride acceptor, were determined by using UV−vis absorption spectroscopy technique. The relationship between the thermodynamic driving forces and kinetic rate of the hydride transfer was examined. Thermodynamic characteristic graph (TCG) of the dihydropyridines as an efficient “Molecule ID Card” was introduced. The TCG can be used to quantitatively diagnose or predict the characteristic chemical properties of the dihydropyridines and their various reaction intermediates. The mechanism of hydride transfer from the dihydropyridines to acridinium perclorate was diagnosed and elucidated by using the determined thermodynamic parameters and the activation parameters.
Co-reporter:Xiao-Qing Zhu and Chun-Hua Wang
The Journal of Physical Chemistry A 2010 Volume 114(Issue 50) pp:13244-13256
Publication Date(Web):November 30, 2010
DOI:10.1021/jp109149x
Combined with the integral equation formalism polarized continuum model (IEFPCM), the hydride affinities of 96 various acylcarbenium ions in the gas phase and CH3CN were estimated by using the B3LYP/6-31+G(d)//B3LYP/6-31+G(d), B3LYP/6-311++G(2df,2p)//B3LYP/6-31+G(d), and BLYP/6-311++G(2df,2p)//B3LYP/6-31+G(d) methods for the first time. The results show that the combination of the BLYP/6-311++G(2df,2p)//B3LYP/6-31+G(d) method and IEFPCM could successfully predict the hydride affinities of arylcarbeniums in MeCN with a precision of about 3 kcal/mol. On the basis of the calculated results from the BLYP method, it can be found that the hydride affinity scale of the 96 arylcarbeniums in MeCN ranges from −130.76 kcal/mol for NO2−PhCH+−CN to −63.02 kcal/mol for p-(Me)2N−PhCH+−N(Me)2, suggesting most of the arylcarbeniums are good hydride acceptors. Examination of the effect of the number of phenyl rings attached to the carbeniums on the hydride affinities shows that the increase of the hydride affinities takes place linearly with increasing number of benzene rings in the arylcarbeniums. Analyzing the effect of the substituents on the hydride affinities of arylcarbeniums indicates that electron-donating groups decrease the hydride affinities and electron-withdrawing groups show the opposite effect. The hydride affinities of arylcarbeniums are linearly dependent on the sum of the Hammett substituent parameters σp+. Inspection of the correlation of the solution-phase hydride affinities with gas-phase hydride affinities and aqueous-phase pKR+ values reveals a remarkably good correspondence of ΔGH−A(R+) with both the gas-phase relative hydride affinities only if the α substituents X have no large electron-donating or -withdrawing properties and the pKR+ values even though the media are dramatically different. The solution-phase hydride affinities also have a linear relationship with the electrophilicity parameter E, and this dependence can certainly serve as one of the most effective ways to estimate the new E values from ΔGH−A(R+) or vice versa. Combining the hydride affinities and the reduction potentials of the arylcarbeniums, we obtained the bond homolytic dissociation Gibbs free energy changes of the C−H bonds in the corresponding hydride adducts in acetonitrile, ΔGHD(RH), and found that the effects of the substituent on ΔGHD(RH) are very small. Simple thermodynamic analytic platforms for the three C−H cleavage modes were constructed. It is evident that the present work would be helpful in understanding the nature of the stabilities of the carbeniums and mechanisms of the hydride transfers between carbeniums and other hydride donors.
Co-reporter:ChaoTun Cao;Hui Liu;Jin-Pei Cheng
Science Bulletin 2010 Volume 55( Issue 25) pp:2799-2802
Publication Date(Web):2010 September
DOI:10.1007/s11434-010-4050-2
A very simple molecular cation, 4-(4-dimethylaminophenyl)-2,6-diphenylpyrylium, has been demonstrated to have a function of molecular half-adder and half-subtractor according to the detectable spectroscopic changes of the molecular system in response to the inputs of acid and base. Distinct algebraic operations can be performed in this reconfigurable molecular logic system.
Co-reporter:LianRui Mei;Jin-Pei Cheng
Science Bulletin 2010 Volume 55( Issue 25) pp:2824-2828
Publication Date(Web):2010 September
DOI:10.1007/s11434-010-3205-5
A polyethylene glycol (PEG)-supported NADH model as a novel organic reductant was designed and synthesized. The reductions of various α, β-unsaturated ketones by the PEG-supported NADH model were examined, and the results showed that the reductions completely and quickly proceed with no catalyst at ambient temperature. The main advantages of this liquid-like PEG-supported NADH model are easy workup, and an optimal potential for recycling use and solvent-free for use in reactions.
Co-reporter:Dorothea Richter Dr.;Yue Tan Dr.;Anna Antipova Dr.;Herbert Mayr Dr.
Chemistry – An Asian Journal 2009 Volume 4( Issue 12) pp:1824-1829
Publication Date(Web):
DOI:10.1002/asia.200900322
Abstract
The rates of the hydride abstractions from the 2-aryl-1,3-dimethyl-benzimidazolines 1a–f by the benzhydrylium tetrafluoroborates 3a–e were determined photometrically by the stopped-flow method in acetonitrile at 20 °C. The reactions follow second-order kinetics, and the corresponding rate constants k2 obey the linear free energy relationship log k2(20 °C)= s(N+E), from which the nucleophile-specific parameters N and s of the 2-arylbenzimidazolines 1a–c have been derived. With nucleophilicity parameters N around 10, they are among the most reactive neutral CH hydride donors which have so far been parameterized. The poor correlation between the rates of the hydride transfer reactions and the corresponding hydricities (ΔH0) indicates variable intrinsic barriers.
Die Geschwindigkeiten der Hydrid-Abstraktionen aus 2-Aryl-1,3-dimethyl-benzimidazolinen 1a–f durch die Benzhydrylium-tetrafluoroborate 3a–e wurden mit der Stopped-Flow-Methode in Acetonitril photometrisch bestimmt. Die Reaktionen verlaufen nach einer Kinetik 2. Ordnung, und die entsprechenden Geschwindigkeitskonstanten k2 befolgen die lineare Freie-Energie-Beziehung log k2(20 °C)=s(N+E), aus der die Nucleophil-spezifischen Parameter N und s der 2-Arylbenzimidazole 1a–c abgeleitet wurden. Mit Nucleophilie-Parametern N von ca. 10 befinden sie sich unter den reaktivsten neutralen C-H Hydriddonoren, die bisher parametrisiert wurden. Die geringe Korrelation zwischen den Geschwindigkeitskonstanten von Hydridtransfer-Reaktionen und den entsprechenden Hydrizitäten (ΔH0) weist auf unterschiedliche intrinsische Barrieren hin.
Co-reporter:Xiao-Qing Zhu, Zhi Dai, Ao Yu, Shuai Wu and Jin-Pei Cheng
The Journal of Physical Chemistry B 2008 Volume 112(Issue 37) pp:11694-11707
Publication Date(Web):August 26, 2008
DOI:10.1021/jp8041268
The thermodynamic driving forces (defined as the enthalpy changes or redox potentials in this work) of the 18 phenothiazines and their analogues, phenoxazine, N-methyl-dihydrophenazine, 9H-thioxanthene, 9H-xanthene and 9,10-dihydro-N-methylacridine, to release hydride, hydrogen atom, proton, and electron in acetonitrile, the thermodynamic driving forces of the radical cations of the phenothiazines and the analogues to release hydrogen atom, proton, and electron in acetonitrile, and the thermodynamic driving forces of the cations of the phenothiazines with two positive charges and their analogues to release proton in acetonitrile were estimated by using experimental methods. The effect of the remote substituents on the 11 determined thermodynamic driving forces were examined according to Brown’s substituent parameters; the results show that the values of the 11 thermodynamic driving forces all are linearly dependent on the sum of Brown substituent parameters (σ+) with very good correlation coefficients, which indicates that for any one- or multisubstituted at para- and/or meta-position phenothiazines and their various reaction intermediates, the 11 thermodynamic driving forces all can be easily and safely estimated from the corresponding Brown substituent parameters (σ+). The relative effective charges on the center nitrogen atom in phenothiazines and their various reaction intermediates were estimated from the related Hammett-type linear free-energy relationships, which can be used to efficiently measure the electrophilicity, nucleophilicity, and dimerizing ability of the corresponding reaction intermediates of phenothiazines and their analogues. All the information disclosed in this work could not only supply a gap of the chemical thermodynamics on the mutual conversions between phenothiazines and their various reaction intermediates in solution but also strongly promote the fast development of the chemistry and application of phenothiazines and their analogues.
Co-reporter:Xiao-Qing Zhu Dr.;Min Zhang;Qiao-Yun Liu;Xiao-Xiao Wang;Jian-Yu Zhang;Jin-Pei Cheng Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 24) pp:
Publication Date(Web):3 MAY 2006
DOI:10.1002/anie.200600536
Choosing a suitable hydride reducing agent and thermodynamic analysis of reduction mechanisms is facilitated by experimental hydride affinities ΔH, which are reported herein for 28 polarized olefins 1 in acetonitrile (see scheme). The method should also be applicable to ketones, aldehydes, and imines.
Co-reporter:Xiao-Qing Zhu Dr.;Min Zhang;Qiao-Yun Liu;Xiao-Xiao Wang;Jian-Yu Zhang;Jin-Pei Cheng Dr.
Angewandte Chemie 2006 Volume 118(Issue 24) pp:
Publication Date(Web):3 MAY 2006
DOI:10.1002/ange.200600536
Die Wahl eines Hydridreagens und die thermodynamische Analyse des Reduktionsmechanismus werden durch die experimentellen Hydridaffinitäten ΔH erleichtert, die für 28 polarisierte Olefine 1 in Acetonitril ermittelt wurden (siehe Schema). Diese Methode sollte auch auf Ketone, Aldehyde und Imine anwendbar sein.
Co-reporter:Xiao-Qing Zhu ;Lei Cao;Yang Liu;Yuan Yang;Jin-Yong Lu;Jian-Shuang Wang;Jin-Pei Cheng
Chemistry - A European Journal 2003 Volume 9(Issue 16) pp:
Publication Date(Web):7 AUG 2003
DOI:10.1002/chem.200304714
Five 1-(p-substituted phenyl)-1,4-dihydronicotinamides (GPNAH-1,4-H2) and five 1-(p-substituted phenyl)-1,2-dihydronicotinamides (GPNAH-1,2-H2) were synthesized, which were used to mimic NAD(P)H coenzyme and its 1,2-dihydroisomer reductions, respectively. When the 1,4-dihydropyridine (GPNAH-1,4-H2) and the 1,2-dihydroisomer (GPNAH-1,2-H2) were treated with p-trifluoromethylbenzylidenemalononitrile (S) as a hydride acceptor, both reactions gave the same products: pyridinium derivative (GPNA+) and carbanion SH− by a hydride one-step transfer. Thermodynamic analysis on the two reactions shows that the hydride transfer from the 1,2-dihydropyridine is much more favorable than the hydride transfer from the corresponding 1,4-dihydroisomer, but the kinetic examination displays that the former reaction is remarkably slower than the latter reaction, which is mainly due to much more negative activation entropy for the former reaction. When the formed pyridinium derivative (GPNA+) was treated with SH−, the major reduced product was the corresponding 1,4-dihydropyridine along with a trace of the 1,2-dihydroisomer. Thermodynamic and kinetic analyses on the hydride transfer from SH− to GPNA+ all suggest that the 4-position on the pyridinium ring in GPNA+ is much easier to accept the hydride than the 2-position, which indicates that when the 1,4-dihydropyridine is used the hydride donor to react with S, the formed pyridinium derivative GPNA+ may return to the 1,4-dihydropyridine by a hydride transfer cycle; but when the 1,2-dihydropyridine is used as the hydride donor, the formed pyridinium derivative can not return to the 1,2-dihydropyridine by the hydride reverse transfer from SH− to GPNA+. These results clearly show that the hydride-transfer cycle is favorable for the 1,4-dihydronicotinamides, but unfavorable for the corresponding 1,2-dihydroisomers.
Co-reporter:Xiao-Qing Zhu ;Hai-Rong Li;Qian Li Dr.;Teng Ai;Jin-Yong Lu;Yuan Yang;Jin-Pei Cheng
Chemistry - A European Journal 2003 Volume 9(Issue 4) pp:
Publication Date(Web):11 FEB 2003
DOI:10.1002/chem.200390108
Heterolytic and homolytic bond dissociation energies of the C4H bonds in ten NADH models (seven 1,4-dihydronicotinamide derivatives, two Hantzsch 1,4-dihydropyridine derivatives, and 9,10-dihydroacridine) and their radical cations in acetonitrile were evaluated by titration calorimetry and electrochemistry, according to the four thermodynamic cycles constructed from the reactions of the NADH models with N,N,N′,N′-tetramethyl-p-phenylenediamine radical cation perchlorate in acetonitrile (note: C9H bond rather than C4H bond for 9,10-dihydroacridine; however, unless specified, the C9H bond will be described as a C4H bond for convenience). The results show that the energetic scales of the heterolytic and homolytic bond dissociation energies of the C4H bonds cover ranges of 64.2–81.1 and 67.9–73.7 kcal mol−1 for the neutral NADH models, respectively, and the energetic scales of the heterolytic and homolytic bond dissociation energies of the (C4H).+ bonds cover ranges of 4.1–9.7 and 31.4–43.5 kcal mol−1 for the radical cations of the NADH models, respectively. Detailed comparison of the two sets of C4H bond dissociation energies in 1-benzyl-1,4-dihydronicotinamide (BNAH), Hantzsch 1,4-dihydropyridine (HEH), and 9,10-dihydroacridine (AcrH2) (as the three most typical NADH models) shows that for BNAH and AcrH2, the heterolytic C4H bond dissociation energies are smaller (by 3.62 kcal mol−1) and larger (by 7.4 kcal mol−1), respectively, than the corresponding homolytic C4H bond dissociation energy. However, for HEH, the heterolytic C4H bond dissociation energy (69.3 kcal mol−1) is very close to the corresponding homolytic C4H bond dissociation energy (69.4 kcal mol−1). These results suggests that the hydride is released more easily than the corresponding hydrogen atom from BNAH and vice versa for AcrH2, and that there are two almost equal possibilities for the hydride and the hydrogen atom transfers from HEH. Examination of the two sets of the (C4H).+ bond dissociation energies shows that the homolytic (C4H).+ bond dissociation energies are much larger than the corresponding heterolytic (C4H).+ bond dissociation energies for the ten NADH models by 23.3–34.4 kcal mol−1; this suggests that if the hydride transfer from the NADH models is initiated by a one-electron transfer, the proton transfer should be more likely to take place than the corresponding hydrogen atom transfer in the second step. In addition, some elusive structural information about the reaction intermediates of the NADH models was obtained by using Hammett-type linear free-energy analysis.
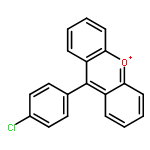
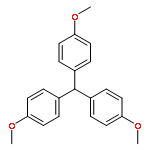
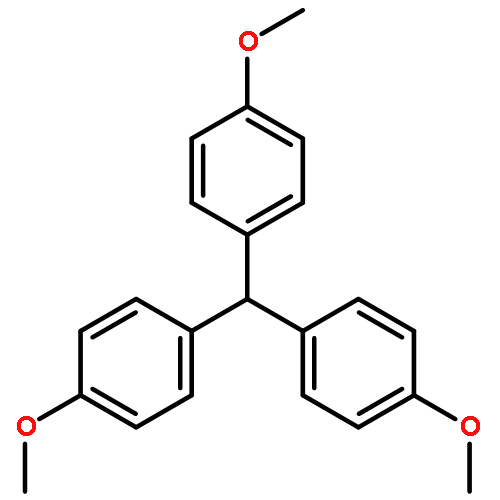
Co-reporter:Fan-kun Meng and Xiao-qing Zhu
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 1) pp:NaN206-206
Publication Date(Web):2016/11/22
DOI:10.1039/C6OB02195F
25 Dihydropyrimidine derivatives, a new class of organo-hydrides, were designed and synthesized by the Biginelli reaction. For the first time, the thermodynamic driving forces of the six elemental steps to obtain a hydride in acetonitrile were determined by isothermal titration and electrochemical methods, respectively. The effects of molecular structures and substituents on these thermodynamic parameters were examined, uncovering some interesting structure–reactivity relationships. Both the thermodynamic and kinetic studies show that the hydride transfer from dihydropyrimidines to 9-phenylxanthylium (PhXn+ClO4−) prefers a concerted mechanism.
Co-reporter:Xiao-Qing Zhu, Fei-Huang Deng, Jin-Dong Yang, Xiu-Tao Li, Qiang Chen, Nan-Ping Lei, Fan-Kun Meng, Xiao-Peng Zhao, Su-Hui Han, Er-Jun Hao and Yuan-Yuan Mu
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 36) pp:NaN6089-6089
Publication Date(Web):2013/07/16
DOI:10.1039/C3OB40831K
A classical but new kinetic equation to estimate activation energies of various hydride transfer reactions was developed according to transition state theory using the Morse-type free energy curves of hydride donors to release a hydride anion and hydride acceptors to capture a hydride anion and by which the activation energies of 187 typical hydride self-exchange reactions and more than thirty thousand hydride cross transfer reactions in acetonitrile were safely estimated in this work. Since the development of the kinetic equation is only on the basis of the related chemical bond changes of the hydride transfer reactants, the kinetic equation should be also suitable for proton transfer reactions, hydrogen atom transfer reactions and all the other chemical reactions involved with breaking and formation of chemical bonds. One of the most important contributions of this work is to have achieved the perfect unity of the kinetic equation and thermodynamic equation for hydride transfer reactions.
Co-reporter:Nan-Ping Lei, Yan-Hua Fu and Xiao-Qing Zhu
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 47) pp:NaN11485-11485
Publication Date(Web):2015/09/29
DOI:10.1039/C5OB01715G
A series of analogues of indazolium alkaloids were designed and synthesized. The thermodynamic driving forces of the 6 elemental steps for the analogues of indazolium alkaloids to obtain hydride in acetonitrile were determined using an isothermal titration calorimeter (ITC) and electrochemical methods, respectively. The effects of molecular structure and substituents on the thermodynamic driving forces of the 6 steps were examined. Meanwhile, the oxidation mechanism of NADH coenzyme by indazolium alkaloids was examined using the chemical mimic method. The result shows that the oxidation of NADH coenzyme by indazolium alkaloids in vivo takes place by one-step concerted hydride transfer mechanism.
Co-reporter:Ke Xia, Guang-Bin Shen and Xiao-Qing Zhu
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 22) pp:NaN6268-6268
Publication Date(Web):2015/04/28
DOI:10.1039/C5OB00538H
32 F420 coenzyme models with alkylation of the three different N atoms (N1, N3 and N10) in the core structure (XFH−) were designed and synthesized and the thermodynamic driving forces (defined in terms of the molar enthalpy changes or the standard redox potentials in this work) of the 32 XFH− releasing hydride ions, hydrogen atoms and electrons, the thermodynamic driving forces of the 32 XFH˙ releasing protons and hydrogen atoms and the thermodynamic driving forces of XF−˙ releasing electrons in acetonitrile were determined using titration calorimetry and electrochemical methods. The effects of the methyl group at N1, N3 and N10 and a negative charge on N1 and N10 atoms on the six thermodynamic driving forces of the F420 coenzyme models and their related reaction intermediates were examined; the results show that seating arrangements of the methyl group and the negative charge have remarkably different effects on the thermodynamic properties of the F420 coenzyme models and their related reaction intermediates. The effects of the substituents at C7 and C8 on the six thermodynamic driving forces of the F420 coenzyme models and their related reaction intermediates were also examined; the results show that the substituents at C7 and C8 have good Hammett linear free energy relationships with the six thermodynamic parameters. Meanwhile, a reasonable determination of possible reactions between members of the F420 family and NADH family in vivo was given according to a thermodynamic analysis platform constructed using the elementary step thermodynamic parameter of F420 coenzyme model 2FH− and NADH model MNAH releasing hydride ions in acetonitrile. The information disclosed in this work can not only fill a gap in the chemical thermodynamics of F420 coenzyme models as a class of very important organic sources of electrons, hydride ions, hydrogen atoms and protons, but also strongly promote the fast development of the chemistry and applications of F420 coenzyme.








![3-[(phenylsulfonyl)methyl]benzonitrile](http://img.cochemist.com/ccimg/51300/51229-58-4.png)
![3-[(phenylsulfonyl)methyl]benzonitrile](http://img.cochemist.com/ccimg/51300/51229-58-4_b.png)












![N-[2-(2,3-DIHYDRO-1,4-BENZODIOXIN-5-YLOXY)ETHYL]-1-PROPANAMINE HY<WBR />DROCHLORIDE (1:1)](http://img.cochemist.com/ccimg/7200/7160-01-2.png)
![N-[2-(2,3-DIHYDRO-1,4-BENZODIOXIN-5-YLOXY)ETHYL]-1-PROPANAMINE HY<WBR />DROCHLORIDE (1:1)](http://img.cochemist.com/ccimg/7200/7160-01-2_b.png)






![Benzonitrile, 4-[(phenylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/51300/51229-59-5.png)
![Benzonitrile, 4-[(phenylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/51300/51229-59-5_b.png)







![Benzenesulfonamide, N-[2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]phenyl]-2,4,6-trimethyl-](/data/chemimg/139800/1019334-58-7.png)
![Benzenesulfonamide, N-[2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]phenyl]-2,4,6-trimethyl-](/data/chemimg/139800/1019334-58-7_b.png)




















![Benzenesulfonamide, N-[2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]phenyl]-4-methyl-](/data/chemimg/139800/1019334-59-8.png)
![Benzenesulfonamide, N-[2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]phenyl]-4-methyl-](/data/chemimg/139800/1019334-59-8_b.png)




![Benzenesulfonamide, N-[2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]phenyl]-4-nitro-](/data/chemimg/139800/1019334-60-1.png)
![Benzenesulfonamide, N-[2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]phenyl]-4-nitro-](/data/chemimg/139800/1019334-60-1_b.png)











![Benzeneacetic acid, α-[(3,4-dimethoxyphenyl)methylene]-3,4-dimethoxy-, methyl ester, (αZ)-](/data/chemimg/103300/172922-64-4.png)
![Benzeneacetic acid, α-[(3,4-dimethoxyphenyl)methylene]-3,4-dimethoxy-, methyl ester, (αZ)-](/data/chemimg/103300/172922-64-4_b.png)
![Benzeneacetic acid, α-[(3,4-dimethoxyphenyl)methylene]-3,4-dimethoxy-, (αZ)-](/data/chemimg/103200/172922-62-2.png)
![Benzeneacetic acid, α-[(3,4-dimethoxyphenyl)methylene]-3,4-dimethoxy-, (αZ)-](/data/chemimg/103200/172922-62-2_b.png)