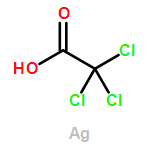
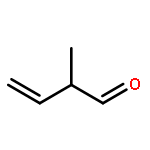
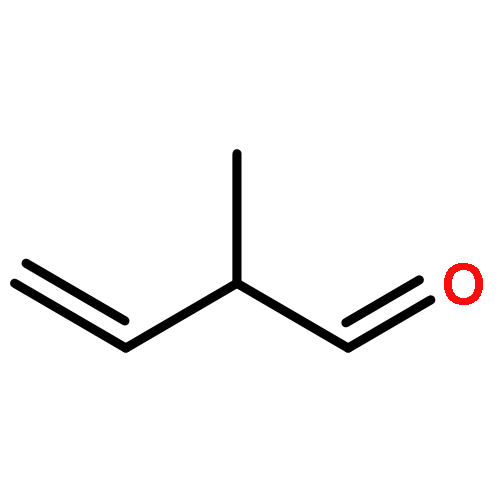
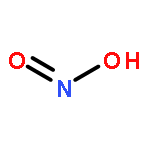
Co-reporter:ZhiFang Xu;Ze Liu;WeiGang Wang
Science Bulletin 2011 Volume 56( Issue 13) pp:1352-1356
Publication Date(Web):2011 May
DOI:10.1007/s11434-011-4461-8
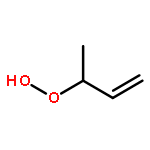
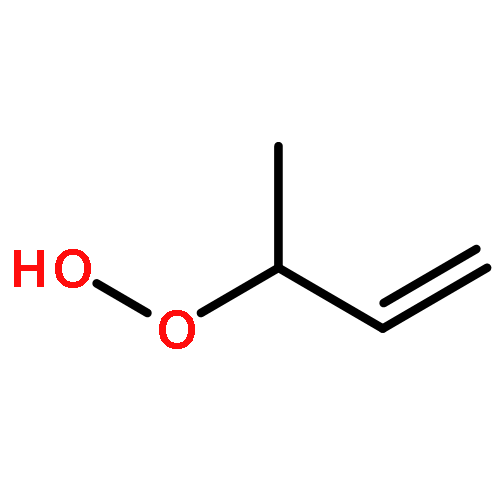
Unsaturated alcohols are important components in complex mixtures of oxygenated volatile organic compounds, and play a significant role in atmospheric chemistry. The uptake kinetics of 3-buten-1-ol (BO31), 4-penten-1-ol (PO41) and 3-methyl-3-buten-1-ol (MBO331) into 20 wt%-80 wt% H2SO4 solutions were studied, using a rotated wetted-wall reactor coupled to a differentially pumped single-photon ionization time of flight mass spectrometer (SPI-TOFMS). With increasing acidity, the uptake processes changed from reversible to irreversible (reactive). Reactive uptake was observed in 60 wt%-80 wt%, 50 wt%-80 wt% and 30 wt%-80 wt% H2SO4 solutions for BO31, PO41 and MBO331, respectively. Reactive uptake coefficients were acquired and are reported here for the first time. Reactivity order followed the trend: BO31
Co-reporter:Shengrui Tong, Chunping Ma, Maofa Ge, Weigang Wang, Dianxun Wang
Journal of Molecular Structure 2010 Volume 978(1–3) pp:108-113
Publication Date(Web):20 August 2010
DOI:10.1016/j.molstruc.2010.02.028
The electronic structure and substituent effects (SEs) in 2-bromo-5-chlorothiophene, 2-bromo-5-methylthiophene, and 2-bromo-5-nitrothiophene have been investigated by HeI photoelectron spectroscopy (PES). The observed PES bands were analyzed by combining empirical arguments and theoretical methods. The outermost electrons of the three compounds all reside mainly in thiophene ring. The analysis of electronic effects of the donor or acceptor substituent groups is essential for the reliable assignment of the observed photoelectron spectra. The investigation of π- and n-orbital ionization potentials has enabled us to describe the substituent effects and the relative reactivities. Furthermore, the natural bond orbital (NBO) analysis was applied for better understanding the nature of the intermolecular interaction.
Co-reporter:Ze Liu, Maofa Ge, Shi Yin, Weigang Wang
Chemical Physics Letters 2010 Volume 491(4–6) pp:146-150
Publication Date(Web):17 May 2010
DOI:10.1016/j.cplett.2010.04.004
Abstract
The uptake and reaction kinetics of α-pinene and β-pinene with H2SO4 solutions were studied over the composition range of 37.0–80.0 wt.% in this Letter. The measurements have identified the occurrence of reversible uptake and irreversible reaction in acid solution. The initial and steady-state uptake coefficients (γi and γs-s) were acquired for the first time, displaying a strong dependence on solution acidity, and β-pinene is more reactive than α-pinene. Atmospheric implication was discussed based on the corresponding uptake coefficients, which demonstrates that this heterogeneous acid-catalyzed reaction might be a significant contributor to SOA loading in concentrated acidic aerosols.
Co-reporter:Ning Zhao;YuanHong Zhang;Xin Liu;XiaoQiang Yu
Science Bulletin 2010 Volume 55( Issue 32) pp:3661-3667
Publication Date(Web):2010 November
DOI:10.1007/s11434-010-4176-2
A new carbazole tricationic salt, 4,4′-(1E,1′E)-2,2′-(9-(2-(1-(2-hydroxyethyl)pyridinium-4-yl)ethyl)-9H-carbazole-3,6-diyl) bis(ethane-2,1-diyl) bis(1-(2-hydroxyethyl)pyridinium) iodide (THEPC) was synthesized. Photophysical experiments have shown that THEPC has large two-photon excited fluorescence action cross-sections (33 GM in the presence of DNA), which ranks THEPC as a good biological fluorophore. The results from electronic absorption, circle dichroism and single-/two-photon fluorescence emission spectra suggest that THEPC can strongly bind to DNA, with an intrinsic binding constant of 5.79 × 106 L mol−1. THEPC has better photostability under one- or two-photon excitation conditions. Finally, the staining photos from two-photon fluorescence microscopy (TPM) show that THEPC can exclusively label the nucleus with high contrast and without image distortion. These remarkable properties and optimized imaging ability make THEPC an attractive DNA probe in TPM.
Co-reporter:ZhiFeng Pu;QianShu Li
Science China Chemistry 2010 Volume 53( Issue 8) pp:1737-1745
Publication Date(Web):2010 August
DOI:10.1007/s11426-010-4037-5
Complexes involving planar octacoordinate alkaline earth metal atoms in the centers of eight-membered boron rings have been investigated by two density functional theory (DFT) methods. BeB82− with D8h symmetry is predicted to be stable, both geometrically and electronically, since a good match is achieved between the size of the central beryllium atom and the eight-membered boron ring. By contrast, the other alkaline earth metal atoms cannot be stabilized in the center of a planar eight-membered boron ring because of their large radii. By following the out-of-plane imaginary vibrational frequency, pyramidal C8v MgB82−, CaB82−, SrB82−, and BaB82− structures are obtained. The presence of delocalized π and σ valence molecular orbitals in D8h BeB82− gives rise to aromaticity, which is reflected by the value of the nucleus-independent chemical shift. The D8h BeB82− structure is confirmed to be the global minimum on the potential energy surface.
Co-reporter:XiaoPeng Wang;ShengRui Tong;WeiGang Wang
Science Bulletin 2010 Volume 55( Issue 35) pp:4018-4025
Publication Date(Web):2010 December
DOI:10.1007/s11434-010-4154-8
The electronic structures of six mono-terpenoids and two of their oxygenated derivatives were studied by He I photoelectron spectroscopy (PES). The observed bands were interpreted on the basis of empirical arguments and theoretical calculations. The first vertical ionization potentials for β-pinene, α-terpinene, terpinolene, γ-terpinene, limonene, myrcene, citral, and terpinene-4-ol were determined to be 8.73, 7.57, 8.26, 8.30, 8.53, 8.68, 8.71, and 8.77 eV, respectively. Most of these values have not been determined by PES before. The correlations of the first vertical ionization potentials of these compounds to the natural logarithms of rate constants for their reactions with the radicals OH, NO3, and O3 were determined. The correlation coefficients for their reactions with OH, NO3, and O3 were 0.97, 0.91, and 0.95, respectively. This method is a powerful technique for predicting the rate constants for the atmospheric oxidation reactions of terpenoids.
Co-reporter:Maofa Ge, Chunping Ma, Shengrui Tong, Wei Xue, Zhifeng Pu and Dianxun Wang
New Journal of Chemistry 2009 vol. 33(Issue 10) pp:2155-2161
Publication Date(Web):28 Jul 2009
DOI:10.1039/B906486A
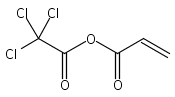
Acryloyl isothiocyanate, CH2CHC(O)NCS, was prepared and studied by IR, Raman, photoelectron spectroscopy (PES), photoionization spectroscopy (PIMS) and theoretical calculations. This molecule was theoretically predicted to prefer the trans-cis (tc) conformation as the most stable conformer, with the CO bond trans to the CC bond and cis to the NCS group. IR and Raman spectra also suggest the presence of the tc conformation only. A theoretical study involving the calculation of the ionization energies using the orbital valence Green’s functional (OVGF/6-311+G(d)) was performed to aid the assignment of the PE spectrum. The first vertical ionization energy of CH2CHC(O)NCS was determined to be 9.89 eV, which is mainly the ionization of the out-of-plane bonding πNCS orbital. Natural population analysis (NPA and NBO) were also performed to investigate the reactivity of CH2CHC(O)NCS.
Co-reporter:Yanbo Gai, Maofa Ge, Weigang Wang
Chemical Physics Letters 2009 Volume 473(1–3) pp:57-60
Publication Date(Web):29 April 2009
DOI:10.1016/j.cplett.2009.03.070
Rate constants for the reaction of ozone with n-butyl acrylate (BUAC) and ethyl methacrylate (ETMEAC) have been investigated for the first time. At 293 ± 1 K and atmospheric pressure, the measured values were (2.40 ± 0.29) × 10−18 cm3 molecule−1 s−1 for BUAC and (7.68 ± 0.88) × 10−18 cm3 molecule−1 s−1 for ETMEAC. The atmospheric lifetimes have also been estimated, which indicate that only in the polluted areas could reaction with ozone be one of the important sinks for these unsaturated esters.Rate constants for the reaction of ozone with n-butyl acrylate (BUAC) and ethyl methacrylate (ETMEAC) at 293 K are determined for the first time using absolute rate method with cyclohexane as OH scavenger.
Co-reporter:Lei Wang, Maofa Ge, Weigang Wang
Chemical Physics Letters 2009 Volume 473(1–3) pp:30-33
Publication Date(Web):29 April 2009
DOI:10.1016/j.cplett.2009.03.047
Kinetic study of the reactions of Cl atoms with EVE and PVE was performed using the absolute rate method over the temperature range 253–298 K at 1 Torr. The obtained Arrhenius expressions were kEVE = (8.0 ± 1.4) × 10−10 exp[(−349 ± 21)/T] cm3 molecule−1 s−1 and kPVE = (1.9 ± 0.3) × 10−9 exp[(−450 ± 18)/T] cm3 molecule−1 s−1. H-atom abstraction is likely to be the main reaction pathway as HCl was detected. The atmospheric lifetimes of EVE and PVE were evaluated, which demonstrates Cl atoms may play an important role for the sink of EVE and PVE.By using a discharge flow-tube system, we determine the rate constants of reactions of Cl atoms with EVE (C2H5OC2H3) and PVE (C3H7OC2H3) under different conditions (253–298 K, 0.5–1.8 Torr). The results demonstrate that reactions of EVE and PVE with Cl atoms may play a significant role for the degradation of EVE and PVE.
Co-reporter:Shi Yin, Wei Xue, Xun-Lei Ding, Wei-Gang Wang, Sheng-Gui He, Mao-Fa Ge
International Journal of Mass Spectrometry 2009 Volume 281(1–2) pp:72-78
Publication Date(Web):15 March 2009
DOI:10.1016/j.ijms.2008.12.014
A time of flight mass spectrometer coupled with a laser ablation/supersonic expansion cluster source is used to study the formation and distribution of cationic iron and cobalt oxide clusters. Although the distributions of iron oxide clusters (FemOnq, q = 0, ±1) have been extensively reported in literature, new and very interesting distribution of FemOn+ clusters is observed in this study. Under saturated O2 growth conditions, the smallest (leading) cluster in m = 2k + 1 (k = 2−14) cluster series is with stoichiometry of Fe2kO3kFeO+, which is perfect (iron atoms are perfectly oxidized) in terms of average oxidation states of iron (Fe3+) and oxygen (O2−) atoms. For m = 2k (k = 2–15) cluster series, the leading cluster is either Fe2kO3k+ (the least over-oxidized) or Fe2kO3k−1+ (the least under-oxidized). Density functional theory (DFT) calculations indicate that these leading clusters are with unexpected structures although their appearance in the mass spectra is predictable. These clusters may serve as good models for predicting or interpreting novel properties of Fe2O3 nano-materials. The distribution of the cobalt oxide clusters (ComOn+) under saturated O2 growth conditions is complex and very different from that of FemOn+. A very interesting result for cobalt species is that two clusters Co11O13+ and Co12O13+ are missing in the cluster distribution although their oxygen-neighbor clusters Co11O12,14+ and Co12O12,14+ are generated. This suggests relatively high stability for Co11O12+ and Co12O12+ clusters. The DFT calculations predict that Co12O12 cluster are with tower or cage structure rather than the compact NaCl-like arrangement that is found for bulk CoO.
Co-reporter:Shengrui Tong, Maofa Ge, Weigang Wang, Carlos O. Della Védova
Journal of Molecular Structure 2009 Volume 919(1–3) pp:83-88
Publication Date(Web):17 February 2009
DOI:10.1016/j.molstruc.2008.08.017
The novel compound fluorocarbonylsulfenyl acetate, FC(O)SOC(O)CH3, which possesses two different carbonyl substituents attached to the SO bond, has been generated through a convenient way by gas–solid reaction between FC(O)SCl and AgOC(O)CH3. The photoelectron and mass spectra of FC(O)SOC(O)CH3 in the gas phase were recorded and assigned. With the combination of experiment, theoretical calculations, and NBO analysis, the electronic and geometrical structures of the title molecule have been investigated. The compound prefers a gauche conformation with both CO bonds syn to the SO bond as the most stable conformation. In FC(O)SOC(O)CH3, the SO bond length and the dihedral angle around SO bond for the most stable conformer are 1.662 Å and 83.0° (B3LYP/6−311++G(3df,3pd)), respectively. The outermost electrons of FC(O)SOC(O)CH3 reside in {35(nS)}−1 orbital, and the experimental first vertical potential of FC(O)SOC(O)CH3 is 10.58 eV.
Co-reporter:Shengrui Tong, Maofa Ge, Weigang Wang, Carlos O. Della Védova
Journal of Molecular Structure 2009 Volume 921(1–3) pp:274-278
Publication Date(Web):17 March 2009
DOI:10.1016/j.molstruc.2009.01.005
The novel compound fluorocarbonylsulfenyl benzoate, FC(O)SOC(O)C6H5, has been generated through a convenient way by gas–solid reaction between FC(O)SCl and AgOC(O)C6H5. The electronic structure of FC(O)SOC(O)C6H5 has been studied by photoelectron spectroscopy (PES), together with quantum chemical calculations. The compound prefers a gauche conformation with both CO bonds syn to the SO bond as the most stable conformation. In FC(O)SOC(O)C6H5, the SO bond length and the dihedral angle around SO bond for the most stable conformer are 1.687 Å and 82.2° (B3LYP/6-311++G(d,p)), respectively. The outermost electrons of FC(O)SOC(O)C6H5 reside in benzyl ring, and the experimental first vertical ionization energy of FC(O)SOC(O)C6H5 is 9.88 eV.
Co-reporter:Maofa Ge, Chunping Ma and Wei Xue
The Journal of Physical Chemistry A 2009 Volume 113(Issue 13) pp:3108-3115
Publication Date(Web):March 5, 2009
DOI:10.1021/jp8110277
Acryloyl isocyanate CH2═CHC(O)NCO is quantitatively prepared by the metathesis reaction between CH2═CHC(O)Cl and AgNCO. Also, jointly with acryloyl chloride, their molecular and electronic structures have been investigated by photoionization mass spectroscopy (PIMS), HeI photoelectron spectroscopy (PES), and theoretical calculations. CH2═CHC(O)NCO was theoretically predicted to prefer the trans−cis (tc) conformation as the most stable conformer, with the C═O bond trans to the C═C bond and cis to the NCO moiety. IR and Raman spectra also suggest the presence of the trans−cis (tc) conformation only. Calculations of the cationic-radical form were carried out in order to compare their properties with those of the neutral molecules. It is worthwhile mentioning that both compounds retain planar structures after ionization. After structural optimizations, a theoretical study involving the calculation of the ionization energies using orbital valence Green’s functional (OVGF) was performed. The ionization energies of different bands in the photoelectron spectrum are in good agreement with the calculated values from the OVGF method. The first vertical ionization energies of CH2═CHC(O)Cl and CH2═CHC(O)NCO are determined to be 10.97 and 10.68 eV, respectively. The HOMOs correspond to the ionization of electrons mainly localized on the πC═C or the πNCO orbitals: {4a′′(πC═C)}−1 and {5a′′(πNCO)}−1, respectively.
Co-reporter:Wei Xue, Shi Yin, Xun-Lei Ding, Sheng-Gui He and Mao-Fa Ge
The Journal of Physical Chemistry A 2009 Volume 113(Issue 18) pp:5302-5309
Publication Date(Web):April 7, 2009
DOI:10.1021/jp810426s
Reactions of small cationic iron oxide clusters (Fe2O4−6+) with N2 are investigated by experiments and first principle calculations. The cationic iron oxide clusters are generated by reaction of laser ablated iron plasma with O2 in a supersonic expansion, and are reacted with N2 in a fast flow reactor at near room temperature conditions. Cluster cations are detected by a time-of-flight mass spectrometer. The substitution reaction Fe2On+ + N2 → Fe2On-2N2+ + O2 is observed for n = 5 but not for n = 4 and 6. Density functional theory calculations predict that the low-lying energy structures of Fe2O4−6+ are with side-on (η1-O2) or end-on (η2-O2) bonded molecular oxygen unit(s). The calculations further predict that the substitution of η1-O2 and η2-O2 in Fe2O4,6+ clusters by N2 is exothermic and subject to negative and positive overall reaction barriers, respectively, at room temperature. We thus propose that the ground state structures of Fe2O4+ and Fe2O6+ contain η2-O2. In contrast, both the experiment and theory favor a η1-O2 in the ground state structure of Fe2O5+.
Co-reporter:ShengRui Tong;WeiGang Wang;ChunPing Ma
Science China Chemistry 2009 Volume 52( Issue 11) pp:
Publication Date(Web):2009 November
DOI:10.1007/s11426-009-0245-2
The electronic structures and substituent effects in o-, m-, and p-iodoanisoles have been investigated by ultraviolet photoelectron spectroscopy (UPS). The observed UPS bands were analyzed by combining empirical arguments and theoretical methods. Owing to the electron-donating nature of both iodo- and methoxy substituents, the first ionization potentials of the three iodoanisoles are lower than those of iodobenzene and anisole. The presence of the two substituents in iodoanisoles leads to an electron- rich structure, which might contribute to the observed high reactivity of iodoanisoles in a number of organic reactions.
Co-reporter:Shengrui Tong;Lin Du;Li Yao;Carlos O. Della Védova
European Journal of Inorganic Chemistry 2008 Volume 2008( Issue 25) pp:3987-3995
Publication Date(Web):
DOI:10.1002/ejic.200800442
Abstract
Fluorocarbonylsulfur thiocyanate, FC(O)SSCN, was generated in the gas phase from a gas–solid reaction of FC(O)SCl on the surface of finely powdered AgSCN. The reaction products were detected and characterized in situ by photoelectron spectroscopy (PES) and photoionization mass spectrometry (PIMS). The geometrical and electronic structures of FC(O)SSCN were investigated by a combination of PES and PIMS experiments, and theoretical calculations. The title compound prefers a gauche conformation with the C=O groups syn to the S–S bond; the structure of the CSSC moiety is characterized by a dihedral angle δCSSC = 89.5° and a bond length rSS = 2.071 Å [at the B3LYP/6-311+G(3df) level] resulting from the sulfur–sulfur lone-pair interactions. After ionization the ground cationic-radical form of FC(O)SSCN·+ adopts a trans-planar structure (δCSSC = 180°) with Cs symmetry. The outermost electrons of FC(O)SSCN reside in the lone pair of the sulfur atom bonded to the C≡N group and the π-bond between the C and N atoms, and the experimental first vertical ionization energy of FC(O)SSCN is 10.89 eV. The possible ionization and dissociation processes of FC(O)SSCN are also discussed.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Maofa Ge;Weigang Wang;Shi Yin;Carlos O. Della Védova
European Journal of Inorganic Chemistry 2008 Volume 2008( Issue 9) pp:1518-1522
Publication Date(Web):
DOI:10.1002/ejic.200701153
Abstract
Oxidovanadium triisocyanate was generated from the heterogeneous reaction of gaseous vanadium trichloride oxide with silver cyanate and studied for the first time in the gas phase. The reaction processes were studied in situ by ultraviolet photoionization mass spectrometry combined with quantum chemical calculations and ultraviolet photoelectron spectrometry. The geometric and electronic structures were characterized and discussed. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Lin Du, Li Yao, Mao-Fa Ge
Journal of Molecular Structure 2008 Volume 882(1–3) pp:146-152
Publication Date(Web):30 June 2008
DOI:10.1016/j.molstruc.2007.09.024
A novel thioperoxide, chlorocarbonylsulfenyl acetate, ClC(O)SOC(O)CH3, has been generated through a convenient way by gas–solid reaction between ClC(O)SCl and AgOC(O)CH3. Photoelectron spectroscopy and theoretical calculations have been performed to investigate the electronic and geometrical structure. ClC(O)SOC(O)CH3 is theoretically predicted to prefer gauche structure as the most stable conformation. In ClC(O)SOC(O)CH3, the S–O bond length and the dihedral angle around S–O bond for the most stable conformer are 1.665 Å and 82.4° (B3LYP/6-311++G(3df,3pd)), respectively. The first vertical ionization energy of ClC(O)SOC(O)CH3 is determined to be 10.43 eV.
Co-reporter:Lin Du, Xiao-Qing Zeng, Mao-Fa Ge, Zheng Sun, Dian-Xun Wang
Journal of Molecular Structure 2008 Volume 878(1–3) pp:26-31
Publication Date(Web):30 April 2008
DOI:10.1016/j.molstruc.2007.07.037
The electronic structures of benzoyl nitrite (C6H5C(O)ONO) and benzoyl nitrate (C6H5C(O)ONO2) have been studied by HeI photoelectron spectroscopy (PES) and quantum chemical calculations. The photoelectron spectra are assigned with the help of the outer valence Green’s function (OVGF) calculations. The first vertical ionization energies of C6H5C(O)ONO and C6H5C(O)ONO2 are determined to be 9.20 and 9.54 eV, respectively. According to the results of theoretical calculations, it can be concluded that a planar CC(O)ONO skeleton in C6H5C(O)ONO and a planar CC(O)ON skeleton in C6H5C(O)ONO2 are the stable structures in the gas phase.
Co-reporter:Mao-Fa GE;Lin DU;Yan-Ping MA;Sheng-Gui HE
Chinese Journal of Chemistry 2008 Volume 26( Issue 6) pp:998-1004
Publication Date(Web):
DOI:10.1002/cjoc.200890211
Abstract
The conformational properties of methanesulfonyl peroxynitrate, CH3S(O)2OONO2 (MSPN), and its radical decomposition products CH3S(O)2OO· and CH3S(O)2O· were studied by ab initio and density functional methods. The dihedral angle around the S–O and the O–O single bond are calculated to be −70.5° and −97.8° (B3LYP/6-311++G(3df,3pd)), respectively. The principal unimolecular dissociation pathways for MSPN were studied using complete basis set (CBS) methods. The reaction enthalpies for the channels CH3S(O)2OONO2 CH3S(O)2OO·+NO2 and CH3S(O)2OONO2CH3S(O)2O·+NO3 were computed to be 111.0 and 140.9 kJ/mol, respectively. The enthalpies of formation at 298 K for MSPN and CH3S(O)2OO radical were predicted to be −358.2 and −281.3 kJ/mol, respectively.
Co-reporter:Mao-Fa GE;Chun-Ping MA
Chinese Journal of Chemistry 2008 Volume 26( Issue 6) pp:983-992
Publication Date(Web):
DOI:10.1002/cjoc.200890209
Abstract
The structures, vibrational spectra, relative energetics, and enthalpies of formation of CH3COIO3 isomers have been investigated with B3LYP, B3P86 and B3PW91 methods in conjugation with the 6-31+G(d), 6-311+G(d,p) and 6-311++G(3df,3pd) basis sets. The CH3COOIO2 structure was found to be the most stable form among the isomers with an estimated enthalpy of formation of −314.6 kJ·mol−1. The enthalpies of formation for CH3COOOOI, CH3COOOIO and CH3COIO3 are −180.7, −184.9 and −50.6 kJ·mol−1, respectively. The implication of the formation of CH3COIO3 isomers from the atmospheric cross-reactions of the acetylperoxy (CH3COO2) and iodine monoxide (IO) radicals was examined and the possible dissociation products of the most likely CH3COIO3 isomers were determined.
Co-reporter:Maofa Ge, Weigang Wang, Shi Yin
Chemical Physics Letters 2008 Volume 453(4–6) pp:296-300
Publication Date(Web):3 March 2008
DOI:10.1016/j.cplett.2008.01.046
We have investigated the heterogeneous interaction between dimethyl sulfide (DMS) and soot surface by a low-pressure flow tube coupled single-photo ionization time of flight mass spectrometry. There are two processes in the DMS loss on soot, the irreversible process is the main process in this experiment. The reversible process takes up small percentage which is only a fraction of less than 20% of the adsorbed DMS. The initial uptake coefficient is 0.013 ± 0.005 at 298 K.By using the coated wall flow tube combined with a differentially pumped single-photo ionization time of flight mass spectrometry, which was designed for the studies on the heterogeneous absorption and reaction, we measured the uptake coefficient of DMS on soot at 298 K in this work and discuss the implication of this result.
Co-reporter:Chun-Ping Ma, Mao-Fa Ge
Journal of Molecular Structure 2008 Volume 891(1–3) pp:221-227
Publication Date(Web):26 November 2008
DOI:10.1016/j.molstruc.2008.03.030
Dimethyl monothiocarbonate, CH3OC(O)SCH3, was synthesized by reaction of methyl chlorothioformate with dried methanol, the product was separated and purified by trap-to-trap condensation before detection and characterization by the photoelectron and photoionization mass spectroscopy. The geometric and electronic properties of CH3OC(O)SCH3 were investigated by the combination of experimental and theoretical studies. The assignment of the bands in the photoelectron spectrum is reasonably supported by previous studies on analogous molecules, as well as the outer-valence Green’s function (OVGF) calculations. The first ionization process happens on the 3p out-of-plane lone pair of sulfur atom n′′S, and the experimental first vertical ionization potential is 9.46 eV. It is worth mentioning that the CSCO dihedral angle twists after ionization and the cationic-radical form becomes non-planar. In the PIMS, it shows six peaks: CH3+, CH3O+, CH3S+, CH3OCO+, CH3SCO+ and CH3OC(O)SCH3+ (M·+), with the dominant features being the CH3SCO+ peak. According to the calculated bond dissociation energies, the dissociation process was discussed. The calculated results indicate that the most preferred dissociation pathway for the parent ion is to form CH3SCO+ and CH3O.
Co-reporter:Chun-Ping Ma, Li Yao, Mao-Fa Ge
Journal of Molecular Structure 2008 Volume 881(1–3) pp:123-131
Publication Date(Web):18 June 2008
DOI:10.1016/j.molstruc.2007.08.034
The electronic structures of a number of halopyridines were measured by HeI photoelectron (PE) spectroscopy. Combined the empirical arguments and theoretical methods, the observed PE bands were interpreted. The careful analysis of measured π-orbital and halogen lone pair ionization energies enable us to describe substituent effects in terms of inductive, resonance, and spin–orbit coupling interactions. The Natural Population Analysis (NPA) has been also used to illuminate the substituent effects between halogen atoms with the parent ring.
Co-reporter:Chun-Ping Ma, Xiao-Qing Zeng, Mao-Fa Ge
Journal of Molecular Structure 2008 Volume 875(1–3) pp:143-151
Publication Date(Web):17 March 2008
DOI:10.1016/j.molstruc.2007.04.020
The HeI photoelectron spectra of acetic anhydride and halogen substituted acetic anhydrides: CH3C(O)OC(O)CH3, CH3C(O)OC(O)CCl3, CH3C(O)OC(O)CF3 and CF3C(O)OC(O)CF3 are obtained and analyzed. Geometry optimizations of the structure for stable conformers are performed at different levels of theory (HF/6-31G(d), B3LYP/6-31G(d), B3LYP/6-311G(d), B3LYP/6-311+G(d)), and the vertical ionization energies are calculated by using orbital valence Green’s functional (OVGF). The experimental first vertical ionization potentials for the four molecules are 10.73, 11.06, 11.53 and 12.21 eV, respectively. The electronic withdrawing effects are clearly reflected in this series of compounds. The highest occupied molecular orbital (HOMO) for each compound is mainly the carbonyl oxygen lone pair (nO) ({13b(nO)}−1, {51a(nO, nCl)}−1, {39a(nO)}−1 and {25b(nO)}−1, respectively). Calculations of the corresponding radical-cationic forms are carried out in order to compare their properties with those of the neutral molecules. All calculations predict the (s, s) conformer is energetically favorable for all molecules investigated, and adopts a planar structure after ionization.
Co-reporter:Lin Du, Li Yao, Mao-Fa Ge, Dian-Xun Wang
Journal of Molecular Structure 2008 Volume 875(1–3) pp:400-405
Publication Date(Web):17 March 2008
DOI:10.1016/j.molstruc.2007.05.012
Trichloromethanesulfenyl benzoate, C6H5C(O)OSCCl3 was prepared in the gas phase through gas–solid heterogeneous reaction between CCl3SCl vapor and powdered C6H5C(O)OAg. The electronic structure of C6H5C(O)OSCCl3 has been studied by photoelectron spectroscopy (PES), together with quantum chemical calculations. The S–O bond length (rSO) and the torsional angle around S–O bond (δCSOC) of the gauche conformer are determined to be 1.691 Å and 104.4° at the MP2/6-311++G(d,p) level, respectively. The first vertical ionization energy of C6H5C(O)OSCCl3 is 9.46 eV, as revealed by PES. The ionization process of the first ionization potential comes from the π orbital at benzene ring.
Co-reporter:Chun-Ping Ma, Wei-Gang Wang, Xiao-Qing Zeng, Mao-Fa Ge
Journal of Molecular Structure 2008 Volume 876(1–3) pp:9-14
Publication Date(Web):30 March 2008
DOI:10.1016/j.molstruc.2007.05.042
FPAN was characterized by photoelectron spectroscopy (PES) and photoionization mass spectrometry (PIMS), the geometry and electronic structures were investigated by combining theoretical calculations. The joint spectroscopic and theoretical studies indicate that FPAN adopts a syn conformer, with the CF3CO and NO2 being a gauge orientation, the cation adopts a planar structure. In PIMS spectrum, the dominant fragment is NO2+ and demonstrates that the O–NO2 bond of FPAN is weak. The outermost electrons of FPAN predominantly localize on the oxygen lone pair of CO, and the experimental first vertical ionization potential of FPAN is 12.36 eV.
Co-reporter:Lin Du, Li Yao, Xiao-Qing Zeng, Mao-Fa Ge
Journal of Molecular Structure 2008 Volume 876(1–3) pp:140-146
Publication Date(Web):30 March 2008
DOI:10.1016/j.molstruc.2007.06.019
Two novel thioperoxides, methoxycarbonylsulfenyl acetate, CH3OC(O)SOC(O)CH3, and methoxycarbonylsulfenyl trifluoroacetate, CH3OC(O)SOC(O)CF3 were generated through heterogeneous reactions and studied by photoelectron spectroscopy (PES) and quantum chemical calculations. The prevailing components in the gas phase may be the gauche (sp–sp–sp) conformers, which exhibit gauche conformation around the SO bond and the CO bonds syn to the SO bond. The torsional angle δCSOC of CH3OC(O)SOC(O)CH3 and CH3OC(O)SOC(O)CF3 are theoretically predicted to be 73.0° and 71.9° (MP2/6-311++G(d,p)), respectively. The first vertical ionization energies of CH3OC(O)SOC(O)CH3 and CH3OC(O)SOC(O)CF3 are determined to be 9.49 and 10.17 eV, respectively. According to the experimental results and theoretical analysis, the HOMOs of these two molecules both correspond to the electrons mainly localized on the sulfur 3p lone pair MOs.
Co-reporter:Shi Yin, Li Yao, Xiao-Qing Zeng, Man-Yu Li, Mao-Fa Ge
Journal of Molecular Structure 2008 Volume 872(Issue 1) pp:24-29
Publication Date(Web):15 January 2008
DOI:10.1016/j.molstruc.2007.02.010
The electronic structures of C4H2Cl2N2 isomers have been studied by HeI photoelectron spectroscopy (PES) combined with the outer valence Green’s function (OVGF) calculations at 6-311++G(d,p) basis sets. The vertical first ionization potentials for 2,6-dichloropyrazine, 2,3-dichloropyrazine, 4,6-dichloropyrimidine and 3,6-dichloropyridazine are determined to be 9.93, 9.89, 10.45 and 10.07 eV, respectively, and the PE spectra were assigned based on molecular orbital analysis and by comparison with related compounds. The effects of chlorine substituent and different positions of nitrogen atoms in these C4H2Cl2N2 isomers have been carefully analyzed by comparing the C4H2Cl2N2 isomers with their matrixes (pyrazine, pyridazine, pyrimidine). The inductive effect of chlorine substituent leads to the stabilization of the nitrogen lone pair orbitals. The resonance effect predominates over the inductive effect, and the consequence is the destabilization of the π orbitals. Owing to the different positions, the effects of nitrogen atoms are different and lead to the different sequence of π orbitals.
Co-reporter:Shi Yin;WeiGang Wang
Science Bulletin 2008 Volume 53( Issue 5) pp:733-738
Publication Date(Web):2008 March
DOI:10.1007/s11434-007-0488-2
The importance of the iodine chemistry in the atmosphere has been demonstrated by recent observations. The uptake of ethyl iodine on black carbon surface was investigated at 298 K for the first time. Degussa FW2 (an amorphous black carbon comprising medium oxides) was used as black carbon sample. Black carbon surface was found to be deactivated in reaction with C2H5I, and the uptake coefficient (γ) was dependent on the time of exposure. The value of (2.3±0.9)×10−2 was determined for the initial uptake coefficient (γ0). The result suggests that the heterogeneous loss of C2H5I on carbonaceous aerosols may be important under the atmospheric conditions.
Co-reporter:Shi Yin;YanPing Ma;Lin Du;ShengGui He
Science Bulletin 2008 Volume 53( Issue 24) pp:3829-3838
Publication Date(Web):2008 December
DOI:10.1007/s11434-008-0502-3
The time of flight mass spectrometer coupled with a laser ablation/supersonic expansion cluster source and a fast flow reactor was adopted to study the reactivity of cationic vanadium oxide clusters (VmOn+) toward acetylene (C2H2) molecules under gas phase (P, ∼ 1.14 kPa), under near room temperature (T, ∼ 350 K) conditions. Association products, VmOnC2H2+ (m,n = 2,4; 2,6; 3,7–8; 4,9–11; 5,12–13; 6,13–16, and 7,17), are observed. The oxidation of C2H2 by (V2O5)n+ (n = 1–3) is experimentally identified. The reactivity of (V2O5)n+ decreases as n increases. Density functional theory (DFT) calculations were carried out to interpret the reaction mechanisms. The DFT results indicate that a terminal oxygen atom from V2O5+ can transfer overall barrierlessly to C2H2 at room temperature, which is in agreement with the experimental observation. Other experimental results such as the observation of V2O6C2H2+ and nonobservation of V2O7,8C2H2+ in the experiments are also well interpreted based on the DFT calculations. The reactivity of vanadium oxide clusters toward acetylene and other hydrocarbons may be considered in identifying molecular level mechanisms for related heterogeneous catalysis.
Co-reporter:Kun Wang;Lin Du
Science Bulletin 2008 Volume 53( Issue 23) pp:3620-3625
Publication Date(Web):2008 December
DOI:10.1007/s11434-008-0537-5
The rate constants for the ozone reactions with n-butyl methyl sulfide (n-BMS, CH3CH2CH2CH2SCH3), sec-butyl methyl sulfide (s-BMS, CH3CH2(CH3)CHSCH3) and tert-butyl methyl sulfide (t-BMS, (CH3)3CSCH3) were measured using our smog chamber under supposedly pseudo-first-order conditions at 300±2 K and 760 Torr. The experimental determined rate constants for n-butyl, s-butyl and t-butyl methyl sulfide are (1.23 ± 0.06)×10−19, (5.08 ± 0.19)×10−20 and (2.26 ± 0.14)×10−20 cm3·molecule−1·s−1, respectively. The reactivity-structure relationship of the reactions was discussed and used to illustrate the mechanism of the ozone reaction with thioethers. The results enrich the kinetics data of atmospheric chemistry.
Co-reporter:Maofa Ge, Li Yao
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2008 Volume 71(Issue 4) pp:1499-1502
Publication Date(Web):15 December 2008
DOI:10.1016/j.saa.2008.05.016
The electronic structures and substituent effects of o-, m-, and p-chloronitrobenzene and bromonitrobenzene have been studied by ultraviolet photoelectron spectroscopy (UPS). It was found that the o-isomer possesses particular electronic properties. This characteristic depends on the conjugation between the benzene ring π orbital and the nitro group π orbital and the interaction of the halogen and nitro groups in the adjacent position.The electronic structures and substituent effects of o-, m-, and p-chloronitrobenzene and bromonitrobenzene have been studied by ultraviolet photoelectron spectroscopy (UPS). It was found that the o-isomer possesses particular electronic properties. This characteristic depends on the conjugation between the benzene ring π orbital and the nitro group π orbital and the interaction of the halogen and nitro groups in the adjacent position.
Co-reporter:Li Yao;Lin Du;Shi Yin
Science China Chemistry 2008 Volume 51( Issue 4) pp:316-321
Publication Date(Web):2008 April
DOI:10.1007/s11426-007-0105-x
A study of the atmospheric photochemical reaction of CF3 radical with CO and O2 was performed by using a homemade ultraviolet photoelectron spectrometer-photoionization mass spectrometer (PES-PIMS). The electronic structures and mechanism of ionization and dissociation of CF3OC(O)OOC(O)-OCF3 were investigated. It was indicated that the two bands on the photoelectron spectrum of CF3OC(O)OOC(O)OCF3 are the result of ionization of an electron from a lone pair of oxygen and a fluorine lone pair of CF3 group. The outermost electrons reside in the oxygen lone pair. The experimental and theoretical first vertical ionization energy is 13.21 and 13.178 eV, respectively, with the PES and OVGF method. They are in good agreement. The photo ionization and dissociation processes were discussed with the help of theoretical calculations and PES-PIMS experiment. After ionization, the parent ions prefer the dissociation of the C—O bond and giving the fragments CF3OCO+ and CF3+. It demonstrated that the ultraviolet photoelectron and photoionization mass spectrometer could be applied widely in the study of atmospheric photochemical reaction.
Co-reporter:Lin Du;Li Yao
European Journal of Inorganic Chemistry 2007 Volume 2007(Issue 28) pp:
Publication Date(Web):3 AUG 2007
DOI:10.1002/ejic.200700455
Two novel dichalcogens, (methoxycarbonyl)sulfenyl thiocyanate, CH3OC(O)SSCN, and (methoxycarbonyl)sulfenyl selenocyanate, CH3OC(O)SSeCN have been generated in a convenient way by gas-solid reactions between CH3OC(O)SCl and AgSCN or AgSeCN. Photoelectron spectroscopy and theoretical calculations have been performed to investigate their electronic and geometrical structures. Both compounds are theoretically predicted to prefer gauche structure as the most stable conformation. In CH3OC(O)SSCN, the S–S bond length and the dihedral angle around the S–S bond for the most stable conformer are 2.075 Å and 80.5°, respectively. And the S–Se bond length and the dihedral angle around the S–Se bond in CH3OC(O)SSeCN are predicted to be 2.210 Å and 80.4°, respectively. The first vertical ionization energies of CH3OC(O)SSCN and CH3OC(O)SSeCN are determined to be 10.19 and 9.84 eV, respectively. The HOMOs correspond to the electron mainly localized on the S 3p or Se 4p lone pair MOs: {38a(nS(SCN))}–1 and {47a(nSe)}–1, respectively. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:Lin Du, Yongfu Xu, Maofa Ge, Long Jia, Li Yao, Weigang Wang
Chemical Physics Letters 2007 Volume 436(1–3) pp:36-40
Publication Date(Web):27 February 2007
DOI:10.1016/j.cplett.2007.01.025
The gas phase reaction of ozone with dimethyl sulfide (DMS) has been studied in our self-made chamber. Experiments were conducted under supposedly pseudo-first-order decay conditions, keeping [DMS]0 > 10[O3]0, but having different combinations of [DMS]0 and [O3]0. Cyclohexane was added into the reactor to eliminate the effect of OH radicals. A value of (1.04 ± 0.21) × 10−19 cm3 molecule−1 s−1 for rate constant was obtained under room temperature of about 301 K. The wall effects and the role of cyclohexane are discussed. Our results enrich the kinetics data of atmospheric chemistry, and further confirm that the gas phase reaction of DMS with ozone is not important for the loss of DMS in the atmosphere.The gas phase reaction of ozone with dimethyl sulfide (DMS) has been studied in our self-made chamber under supposedly pseudo-first-order decay conditions. A value of (1.04 ± 0.21) × 10−19 cm3 molecule−1 s−1 for rate constant was obtained under room temperature of about 301 K, which enriched the kinetic data of atmospheric chemistry.
Co-reporter:Li Yao, Xiao-Qing Zeng, Mao-Fa Ge, Dian-Xun Wang
Journal of Molecular Structure 2007 Volume 841(1–3) pp:104-109
Publication Date(Web):30 September 2007
DOI:10.1016/j.molstruc.2006.11.069
The electronic structures of trisubstituted boroxine Me3B3O3 and (MeO)3B3O3 were investigated by using photoelectron spectroscopy. Ab initio and DFT calculations have been carried out for the assignments of the PE spectra. The experimental first ionization energies of Me3B3O3 and (MeO)3B3O3 are 11.42 and 10.78 eV. These correspond to the ionization of the electron from the boron–carbon σ bonding orbital and the lone pair π orbital on the oxygen atom out of the boroxine ring, respectively.
Co-reporter:Lin Du;YongFu Xu;Long Jia;Li Yao
Science Bulletin 2007 Volume 52( Issue 12) pp:1629-1634
Publication Date(Web):2007 June
DOI:10.1007/s11434-007-0243-8
Large quantities of di-tert-butyl peroxide (DTBP) have been emitted into the troposphere due to human activities. Its role in the atmospheric photochemical reaction has not been understood. This study presents the results of the photochemical reactions of DTBP and NOx, which have been simulated in a self-made smog chamber under the temperature of (29±1)°C. Both the wall decays of ozone and NO2 could be neglected, compared to the results in simulative experiments. The effective intensity of UV light used in the experiments was 1.28×10−3 s−1, which was expressed by the rate constant of NO2 photolysis in purified air. The reaction mechanism was proposed according to our results and reports of other researchers. The maximum values of incremental reactivity (IR) in the three simulative experiments were 9.53×10−2, 5.23×10−2 and 3.78×10−2, respectively. The incremental reactivity decreased with the increase of initial concentrations of DTBP. The IR value of DTBP obtained in this study was comparable to that of acetylene reported in our previous research.
Co-reporter:WeiGang Wang;Li Yao;XiaoQing Zeng;ZiFa Wang
Science Bulletin 2007 Volume 52( Issue 22) pp:3056-3060
Publication Date(Web):2007 November
DOI:10.1007/s11434-007-0474-8
The short-lived reactive specimen nitrous acid HONO was generated in the gas phase by the heterogeneous reaction of gaseous HCI with AgNO2 which can generate higher concentration of HONO than other methods. We investigated the process from generation to dissociation in the gas phase under different controlled temperatures, and discussed the ionization and reaction on the solid surface by combination of the photoelectron spectroscopy and photoionization mass spectroscopy (PES-PIMS) and in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS).
Co-reporter:Li Yao;Xiaoqing Zeng;Weigang Wang;Zheng Sun;Lin Du;Dianxun Wang
European Journal of Inorganic Chemistry 2006 Volume 2006(Issue 12) pp:
Publication Date(Web):18 APR 2006
DOI:10.1002/ejic.200600163
The unstable nitrosyl thiocyanate molecule has been generated in the gas phase for the first time from an in situ heterogeneous reaction at low temperature. The product was detected and characterized by a photoelectron spectrometer-photoionization mass spectrometer (PES-PIMS). The electronic and geometric structures of the molecule were investigated with the help of quantum chemical calculations at the B3LYP, CBS-QB3, and CCSD(T) levels. The joint spectroscopic and theoretical studies provided evidence for the formation of nitrosyl thiocyanate, and indicated that the molecule adopts an open-chain, bent structure with the NO and SCN groups bonding by a relatively strong interaction. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Zeng Xiaoqing;Wang Weigang;Liu Fengyi;Ge Maofa;Sun Zheng;Wang Dianxun
European Journal of Inorganic Chemistry 2006 Volume 2006(Issue 2) pp:
Publication Date(Web):9 DEC 2005
DOI:10.1002/ejic.200500720
Two highly explosive binary triazides of the group 15 elements P(N3)3 and As(N3)3 have been obtained in the gas phase through the heterogeneous reaction of PCl3 and AsCl3, respectively with AgN3 at room temperature. The electronic structures of both triazides have been characterized by photoelectron spectroscopy, combined with quantum chemical calculations. This represents the first electronic study of covalent triazides. The first experimental vertical ionization potentials for P(N3)3 and As(N3)3 are 9.74 and 9.98 eV, with the contribution primarily from the lone pairs of the azido moiety and the arsenic atom, respectively. The results indicate the relative “isolation” of azido moieties in triazides and less stability of these highly explosive compounds in comparison to monoazides and diazides. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Weigang Wang, Maofa Ge, Dianxun Wang
Chemical Physics 2006 Volume 328(1–3) pp:165-172
Publication Date(Web):29 September 2006
DOI:10.1016/j.chemphys.2006.06.034
Abstract
The calculations of geometric structures, relative energies, vibrational frequencies, infrared intensities and binding energies of OIO–H2O and OIO–2H2O clusters have been performed using three DFT methods (B3LYP, B3P86 and B3PW91) at 6-311++G(3df, 3pd) basis set level. There are two kinds of interactions between iodine dioxide and water, one is the hydrogen bonding between the oxygen atom on iodine dioxide and the hydrogen atom on water, another is van der Waals interaction between I atom and the oxygen atom on H2O. The analysis of the natural bond orbital (NBO) second-order interaction energies has also been employed to illuminate the binding energies and the stability of these OIO–nH2O (n = 1, 2) complexes.
Co-reporter:Li Yao;Mao-Fa Ge;Dian-Xun Wang;Cheng-Yin Wu;Nan Xu;Qi-Huang Gong
Chinese Journal of Chemistry 2006 Volume 24(Issue 7) pp:
Publication Date(Web):4 JUL 2006
DOI:10.1002/cjoc.200690165
Ionization and dissociation of nitrosyl chloride ClNO were studied using femtosecond laser mass spectra technique. Strong fragmental ions NO+and Cl+were observed with the laser intensity varied from 3.2×1014 to 2.5×1015 W/cm2. These fragmental ions were attributed to the direct dissociation of the parent ions. Electronic structure calculations were also carried out with Hartree-Fock, density functional and correlated levels of theory to understand the possible fragmentation pathways. The very low N–Cl bond energy in the parent ion of nitrosyl chloride is a clear reason for the absence of ClNO+ and ClN+ ion peaks from the femtosecond laser mass spectrum.
Co-reporter:Li Yao, Xiaoqing Zeng, Maofa Ge, Yunfeng Ding, Weigang Wang, Lin Du, Zheng Sun, Qiao Sun, Dianxun Wang
Chemical Physics Letters 2006 Volume 422(4–6) pp:466-469
Publication Date(Web):10 May 2006
DOI:10.1016/j.cplett.2006.03.028
Abstract
A continuous beam of isopropylthio radical (CH3)2CHS was generated in the gas phase by the pyrolysis of isopropyl disulfide C3H7SSC3H7. The electronic structure of isopropylthio radical has been investigated by the in situ HeI photoelectron spectroscopy, in combination with density functional theory and G2 calculations. The first PE band at 8.95 eV with vibrational progression of 625 ± 60 cm−1 is designated as the X3A″ ground ionic state of (CH3)2CHS+, deriving from ionization of the electron from the SHOMO. Both photoelectron spectroscopy experiment and theoretical calculations provide evidence for the existence of different ionic states of isopropylthio radical for the first time.
Co-reporter:Long Jia, Yongfu Xu, Maofa Ge, Lin Du, Gengchen Wang, Guoshun Zhuang
Acta Physico-Chimica Sinica 2006 Volume 22(Issue 10) pp:1260-1266
Publication Date(Web):October 2006
DOI:10.1016/S1872-1508(06)60060-0
Kinetics of the reaction of ozone with propylene under real atmospheric environmental conditions with an ozone concentration of ca 6.6×10−8 has been investigated in a self-made Teflon Chamber. Using Model 49C-O3 Analyzer and GC-FID, reaction rate constants at a temperature range of 282–314 K were determined by an absolute rate technique in terms of measurements of ozone concentrations. Results show that the reaction rate constant is 6.73×10−18 cm3·molecule−1·s−1 for the initial ozone concentration of 6.61×10−8 and the temperature of 282 K. According to the reaction rate constants under different temperatures, the Arrhenius equation of k2=(5.8±1.2)×10−15e(−1907±53)/T is obtained. Compared with the results reported by other researchers, although the rate constants obtained in this study are systematically underestimated and the activation energy of the reaction overestimated, the results are satisfactory. For example, the largest relative error is only 11% and 5% for the rate constant and activation energy, respectively. These demonstrate that the research equipment used in this study is reliable under real atmospheric conditions and can be used to do further studies related to ozone reactions.
Co-reporter:Weigang Wang Dr. ;Xiaoqing Zeng Dr.;Dianxun Wang ;Li Yao Dr.;Zheng Sun Dr.
ChemPhysChem 2006 Volume 7(Issue 6) pp:1382-1387
Publication Date(Web):8 MAY 2006
DOI:10.1002/cphc.200600084
Sulfur diisocyanate is generated from a heterogeneous reaction of gaseous sulfur dichloride with silver cyanate and studied for the first time in the gas phase. Combined with quantum chemical calculations, the electronic structure is characterized by photoelectron spectroscopy (PES). Simultaneously, an investigation of the possible ionization and dissociation processes for the molecular cation is presented based on experimental soft ionization mass spectrometry. From the calculated bond-dissociation energies, the dissociation pathway is determined. S(NCO)2+ undergoes 1,3-sigmatropic rearrangement with a smaller barrier height (9.9 kcal mol−1) than the neutral counterpart. Thus, the 1,3-sigmatropic rearrangement is preferred for the molecular cation, and OCNCO+ and NS+ is produced by subsequent dissociation of the rearrangement product. The analysis agrees very well with the experimental mass spectrum.
Co-reporter:Xiaoqing Zeng, Li Yao, Weigang Wang, Fengyi Liu, Qiao Sun, Maofa Ge, Zheng Sun, Jianping Zhang, Dianxun Wang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2006 Volume 64(Issue 4) pp:949-955
Publication Date(Web):July 2006
DOI:10.1016/j.saa.2005.09.003
The gas phase electronic structures of CM3C(O)ONO and CM3C(O)ONO2 (M = H, Cl, F) are studied by photoelectron spectroscopy (PES) combined with the outer valence Green's function (OVGF) calculations at 6-311 + G(d, p) basis sets. The highest occupied molecular orbital (HOMO) for each compound is the carbonyl oxygen lone pair (nO), the ionizations of these orbitals are associated with the vibrational frequency about 1750 and 1820 cm−1 reflected on the first band, respectively, for acyl nitrites and nitrate. Comparing with the calculated energies, it can be concluded that the syn conformers with Cs overall symmetry, a planar CC(O)ONO skeleton in nitrites, and a planar CC(O)ON skeleton in nitrates, respectively, are the most stable in the gas phase.
Co-reporter:Sun Qiao;Li Zhen;Zeng Xiao-Qing;Ge Mao-Fa;Wang Dian-Xu
Chinese Journal of Chemistry 2005 Volume 23(Issue 5) pp:
Publication Date(Web):14 JUN 2005
DOI:10.1002/cjoc.200590483
The structural properties of two BrOH2O (1 and 2) and three HOBrH2O complexes (3, 4 and 5) have been investigated using four methods at the 6-311++G(d,p) basis set level. In the two BrOH2O complexes, the complex 2 with 2A′ state, in which the interaction exists between Br atom of BrO and O atom of water, has a binding energies of about 11.37–13.92 J/mol and it is global minimum. As to HOBrH2O complexes, the binding energies of 3 and 4 are about 16.30–21.32 J/mol and the stability order of the three HOBrH2O complexes is: complex 3≈complex 4>complex 5.
Co-reporter:Li Zhou, Weigang Wang, Maofa Ge, Shengrui Tong
Journal of Environmental Sciences (February 2016) Volume 40() pp:44-50
Publication Date(Web):1 February 2016
DOI:10.1016/j.jes.2015.08.018
The heterogeneous uptake processes of hydrogen peroxide on Arizona test dust and two types of authentic Chinese mineral dusts, i.e., Inner Mongolia desert dust and Xinjiang calciferous dust, were investigated using a Knudsen cell reactor coupled with a quadrupole mass spectrometer. The uptake coefficients were measured as a function of the initial concentration of H2O2 from 2.6 × 1011 to 1.2 × 1012 molecules/cm3, and the temperature dependence of the uptake coefficients was investigated over a range from 253 to 313 K. The concentration of H2O2 showed little effect on the uptake coefficients of these heterogeneous processes. As a function of temperature, the initial uptake coefficients decrease with increasing temperature, whereas the steady state uptake coefficients of Arizona test dust and Inner Mongolia desert dust increase with increasing temperature. Implications for the understanding of the uptake processes onto mineral dust samples were also discussed.Download full-size image
Co-reporter:Li Zhou, Weigang Wang, Siqi Hou, Shengrui Tong, Maofa Ge
Journal of Environmental Sciences (December 2015) Volume 38() pp:110-118
Publication Date(Web):1 December 2015
DOI:10.1016/j.jes.2015.05.017
Mineral dust is one of the major aerosols in the atmosphere. To assess its impact on trace atmospheric gases, in this work we present a laboratory study of the effect of temperature on the heterogeneous reaction of NO2 on the surface of ambient Chinese dust over the temperature range from 258 to 313 K. The results suggest that nitrogen dioxide could mainly be adsorbed on these types of Chinese mineral dust reversibly with little temperature dependence. Similar to a previous study on NO2 uptake on mineral aerosols, the uptake coefficients are mainly on the order of 10− 6 for the Chinese dust, when BET areas are taken into account. HONO was observed as a product, and its formation and decomposition on Chinese mineral dust during the uptake processes were also studied. The complete dataset from this study was compiled with previous literature determinations. Atmospheric implications of the heterogeneous reaction between NO2 and mineral dust are also discussed, in an effort to understand this important heterogeneous process.Download full-size image
Co-reporter:Xiaoqing Zeng, Maofa Ge, Zheng Sun, Jiang Bian, Dianxun Wang
Journal of Molecular Structure (17 September 2007) Volume 840(Issues 1–3) pp:
Publication Date(Web):17 September 2007
DOI:10.1016/j.molstruc.2006.11.034
Gaseous nitryl azide N4O2 is generated by the heterogeneous reaction of gaseous ClNO2 with freshly prepared AgN3 at −50 °C. The geometric and electronic structure of the molecule in the gas phase has been characterized by in situ photoelectron spectroscopy (PES) and quantum chemical calculations. The experimental first vertical ionization energy of N4O2 is 11.39 eV, corresponding to the ionization of an electron on the highest occupied molecular orbital (HOMO) {4a″(πnb(N4–N5–N6))}−1. An apparent vibrational spacing of 1600 ± 60 cm−1 (νasO1N2O3) on the second band at 12.52 eV (πnb(O1–N2–O3)) further confirms the preference of energetically stable chain structure in the gas phase. To complement the experimental results, the potential-energy surface of this structurally novel transient molecule is discussed. Both calculations and spectroscopic results suggest that the molecule adopts a trans-planar chain structure, and a five-membered ring decomposition pathway is more favorable.
Co-reporter:Qifan Liu, Weigang Wang, Maofa Ge
Journal of Environmental Sciences (1 May 2015) Volume 31() pp:89-97
Publication Date(Web):1 May 2015
DOI:10.1016/j.jes.2014.09.039
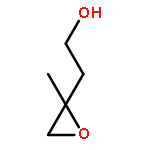
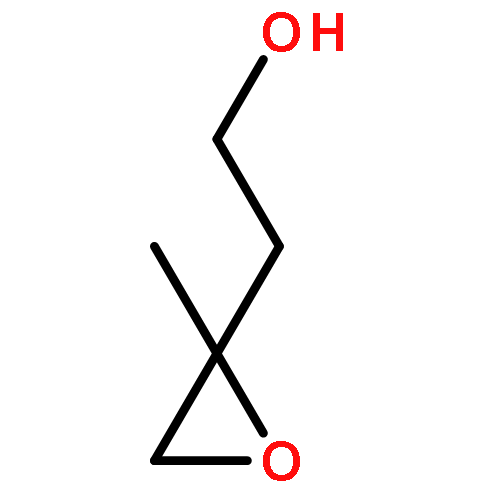
Acid-catalyzed heterogeneous oxidation with hydrogen peroxide (H2O2) has been suggested to be a potential pathway for secondary organic aerosol (SOA) formation from isoprene and its oxidation products. However, knowledge of the chemical mechanism and kinetics for this process is still incomplete. 3-Methyl-2-buten-1-ol (MBO321), an aliphatic alcohol structurally similar to isoprene, is emitted by pine forests and widely used in the manufacturing industries. Herein the uptake of MBO321 into H2SO4–H2O2 mixed solution was investigated using a flow-tube reactor coupled to a mass spectrometer. The reactive uptake coefficients (γ) were acquired for the first time and were found to increase rapidly with increasing acid concentration. Corresponding aqueous-phase reactions were performed to further study the mechanism of this acid-catalyzed reaction. MBO321 could convert to 2-methyl-3-buten-2-ol (MBO232) and yield isoprene in acidic media. Organic hydroperoxides (ROOHs) were found to be generated through the acid-catalyzed route, which could undergo a rearrangement reaction and result in the formation of acetone and acetaldehyde. Organosulfates, which have been proposed to be SOA tracer compounds in the atmosphere, were also produced during the oxidation process. These results suggest that the heterogeneous acid-catalyzed reaction of MBO321 with H2O2 may contribute to SOA mass under certain atmospheric conditions.Download full-size image
Co-reporter:Mengya Lin, Xiaolin Yu, Xueqin Yang, Kezhi Li, Maofa Ge and Junhua Li
Catalysis Science & Technology (2011-Present) 2017 - vol. 7(Issue 7) pp:NaN1580-1580
Publication Date(Web):2017/03/10
DOI:10.1039/C7CY00154A
The exploration of active interfaces has attracted wide attention, especially in the field of catalysis. In this work, Ni/Fe layered double oxide supported Pt nanoparticles (Pt/LDO(N)) were prepared using a hydrothermal and colloid-impregnation method. The Pt/LDO(N) catalyst exhibited remarkable long-term catalytic stability and activity for HCHO oxidation compared with Pt/Fe2O3 and Pt/NiO. The Pt species were well dispersed on the LDO support, and strongly interacted with Fe and Ni by forming an active Pt–Fe/Ni interface. O2 dissociation could happen at the active interface by creating coordinatively unsaturated iron sites, further providing adequate O−/O2− species to take part in HCHO oxidation. The in situ DRIFTS results indicated that the dioxymethylene and formate species were the main reaction intermediates, which could be further oxidized into CO2 and H2O through the involvement of active oxygen.
Co-reporter:Xueqin Yang, Xiaolin Yu, Mengya Lin, Maofa Ge, Yao Zhao and Fuyi Wang
Journal of Materials Chemistry A 2017 - vol. 5(Issue 26) pp:NaN13806-13806
Publication Date(Web):2017/05/31
DOI:10.1039/C7TA03888G
A series of high-efficiency Pt/ZrO2 catalysts were successfully prepared by simple methods on the basis of a ZrO2 support with a mixed monoclinic/tetragonal phase structure. The activity test results showed that the mixed phase catalysts exhibited higher catalytic activity than the pure monoclinic phase, and HCHO can be completely oxidized into CO2 and H2O at near ambient temperature. XRD, Raman and HRTEM results demonstrated that the monoclinic–tetragonal phase interface with abundant defects was formed due to the introduction of the tetragonal phase. According to the results of TEM, XPS and H2-TPR, the mixed phase interfacial structure can induce the formation of the active oxygen species, ionic Ptδ+ species, strong metal-support interaction and low-temperature reducibility, which was vital for the significant improvement of the catalytic activity. Furthermore, the specific HCHO reaction rate of the catalysts at 55 °C increased from 0.8 × 10−3 to 10.4 × 10−3 mmol h−1 m−2 and the activation energy decreased remarkably from 213.5 to 24.7 kJ mol−1 with the increase of the biphase interface content. In situ DRIFTS spectra showed that the special interfacial structure can change the reaction pathway of HCHO oxidation and inhibit the formation of inert carbonate species, thus greatly enhancing the HCHO oxidation activity.
Co-reporter:Ze Liu, Maofa Ge, Weigang Wang, Shi Yin and Shengrui Tong
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 6) pp:NaN2075-2075
Publication Date(Web):2011/01/04
DOI:10.1039/C0CP00905A
Multiphase acid-catalyzed oxidation with hydrogen peroxide (H2O2) has been suggested recently to be a potential route to SOA formation from isoprene and its gas-phase oxidation products, the kinetics and chemical mechanism of this process have not been well-known yet. In this work, the uptake of 2-methyl-3-buten-2-ol (MBO), an important biogenic hydrocarbon and structurally similar to isoprene, into aqueous mixed solutions of H2O2 and sulfuric acid (H2SO4) was performed using a rotated wetted-wall reactor coupled to a differentially pumped single-photon ionization time of flight mass spectrometer (RWW-SPI-TOFMS). The reactive uptake coefficients (γ) were acquired for the first time and the reaction pathways were deduced according to products information. The reactive uptake coefficients of MBO into H2SO4–H2O2 mixed solutions are much greater than that into H2SO4 solutions. Acetaldehyde, acetone and an on-line product, which transformed to isoprene readily in the duration of an off-line experiment, were suggested as products in this process. The further reactions of the carbonyl products can occur in acidic solution, which may play a role in SOA formation. Additionally, in real atmosphere the on-line product is apt to transform to isoprene, an acknowledged precursor of biogenic SOA. Thus, the multiphase acid-catalyzed oxidation of MBO with H2O2 might be a potential contributor to SOA loading.
Co-reporter:Jing Wang, Li Zhou, Weigang Wang and Maofa Ge
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 18) pp:NaN12012-12012
Publication Date(Web):2015/03/30
DOI:10.1039/C4CP05461J
The rate constants and products for the reactions of atomic Cl and O3 molecule with 3-methyl-3-buten-2-one (MBO332) and 3-methyl-3-penten-2-one (MPO332) were determined in a 100 L Teflon chamber at 293 ± 1 K and atmospheric pressure. For MBO332 and MPO332, the rate constants measured with atomic Cl were (2.38 ± 0.26) × 10−10 and (3.00 ± 0.34) × 10−10 cm3 molecule−1 s−1 using the relative rate method. Using the absolute rate method, the rate constants with O3 measured were (1.18 ± 0.21) × 10−17 and (4.07 ± 0.45) × 10−17 cm3 molecule−1 s−1. The products of these reactions were investigated by the proton-transfer-reaction mass spectrum (PTR-MS). The results indicated that the major products observed in the atomic Cl reaction were formaldehyde together with chloroacetone for MBO332, and acetaldehyde and CH3C(O)C(O)Cl for MPO332. For O3 reactions, butanedione and formaldehyde were the main products of MBO332, while butanedione and acetaldehyde were the main products of MPO332. Possible reaction mechanisms were proposed and discussed and the atmospheric implications of these reactions were also discussed.
Co-reporter:Xiaolin Yu, Shengrui Tong, Maofa Ge, Junchao Zuo, Changyan Cao and Weiguo Song
Journal of Materials Chemistry A 2013 - vol. 1(Issue 3) pp:NaN965-965
Publication Date(Web):2012/11/08
DOI:10.1039/C2TA00315E
Composite materials, containing magnetic nanoparticles and cellulose, were synthesized by one-step co-precipitation using NaOH–thiourea–urea aqueous solution for cellulose dissolution. The NaOH in cellulose solution acted as the precipitant of iron oxide nanoparticles, and low-cost cellulose was used as the template to promote the growing of nanoparticles in the cellulose matrix. The method provided a facile, “green” pathway for the fabrication of magnetic nanomaterials. The synthesized cellulose@iron oxide nanoparticles were characterized by FTIR, XRD, SEM, TEM, XPS, TG and VSM. The FTIR, XRD and XPS results demonstrated the formation of Fe2O3 nanoparticles in the composite materials after the co-precipitation. SEM and TEM characterization showed that the Fe2O3 nanoparticles were dispersed in the cellulose matrix due to the synergistic effect. Magnetometric measurements revealed that the resultant composites of cellulose@Fe2O3 nanoparticles exhibited a sensitive magnetic-induced behavior and could be easily separated from aqueous solution through the external magnetic field. The composite materials were applied to remove arsenic from aqueous solution. The results showed that the magnetic nanoparticle composites displayed excellent adsorption efficiency of arsenic compared with other magnetic materials reported, and the Langmuir adsorption capacities of the composites for the removal of arsenite and arsenate were 23.16 and 32.11 mg g−1, respectively.