Co-reporter:Mingming Yu;Zhiwei Gao;Xiaojian Dai;Hui Gong;Lianshan Zhang;Xiaoyan Chen;Da-Fang Zhong;Sherwin K. B. Sy
Clinical Pharmacokinetics 2017 Volume 56( Issue 1) pp:65-76
Publication Date(Web):2017/01/01
DOI:10.1007/s40262-016-0427-y
Apatinib is an oral tyrosine kinase inhibitor approved in China for the treatment of patients with advanced metastatic gastric cancer. The approved dosing schedule is 850 mg once daily. The objective of this study was to develop a population pharmacokinetic (popPK) model of apatinib and determine factors that affect its pharmacokinetics.A popPK model for apatinib was developed using data from 106 individuals, including healthy volunteers and patients with malignant solid tumors. The potential influence of demographic, patient, and laboratory characteristics on oral apatinib pharmacokinetics were investigated in a covariate analysis. The extent of the impact of significant covariates on the exposure of apatinib was evaluated using simulations.The final popPK model was a two-compartment model with mixed first- and zero-order absorption and first-order elimination. The population estimates of apparent clearance (CL/F) and apparent volume at steady-state were 57.8 L/h and 112.5 L, respectively. The non-linear dose proportionality in apatinib relative bioavailability was characterized by a sigmoidal maximum effect (Emax) equation wherein the midpoint dose for the decrease in bioavailability was 766 mg. Patients with advanced gastric cancer exhibited lower bioavailability. Cancer patients in general had lower CL/F than healthy volunteers. Simulation results indicated that apatinib exposure in various population groups were impacted by disease and laboratory characteristics.The increase in apatinib exposure was less than proportional to dose. The pharmacokinetics of apatinib in gastric cancer patients were significantly different from those in patients with other cancer types. Dosing of apatinib in various cancer subpopulations may require adjustments to optimize efficacy and benefits to patients.
Co-reporter:Xin Li, Changyong Yang, Hong Wan, Ge Zhang, Jun Feng, Lei Zhang, Xiaoyan Chen, Dafang Zhong, Liguang Lou, Weikang Tao, Lianshan Zhang
European Journal of Pharmaceutical Sciences 2017 Volume 110(Volume 110) pp:
Publication Date(Web):15 December 2017
DOI:10.1016/j.ejps.2017.01.021
The discovery and development of a novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor SHR1258 (pyrotinib) for the treatment of HER2-postive breast cancer is presented. The structure-activity relationship of lead series and their pharmacokinetic properties were evaluated to identify the potential candidates for further in vivo efficacy studies and preclinical safety assessments. Metabolic pathway and drug-drug interaction were also investigated in preclinical settings. In particular, major metabolites in human and animal species were assessed with regard to potential toxicity or off-target side effects. Overall, the potent and selective EGFR/HER2 dual inhibitor, pyrotinib, displayed robust anti-tumor effects on HER2-overexpressing xenograft models and sufficiently safety windows in animals as well as favorable pharmacokinetic properties in human, which substantially ensures current clinical development. Finally, recent advances of pyrotinib in clinical studies are highlighted with very encouraging outcomes in patients with HER2-postive advanced breast cancer.Download high-res image (224KB)Download full-size image
Co-reporter:Yunting Zhu, Liang Li, Pan Deng, Xiaoyan Chen, Dafang Zhong
Journal of Pharmaceutical and Biomedical Analysis 2016 Volume 117() pp:217-226
Publication Date(Web):5 January 2016
DOI:10.1016/j.jpba.2015.09.001
•TPN729 was a novel phosphodiesterase type 5 inhibitor to treat erectile dysfunction.•Metabolism of TPN729 in humans was first investigated using UPLC/Q–TOF MS.•A total of 22 metabolites were detected and identified.•Seven metabolites were confirmed by comparison with the reference substances.•TPN729 mainly underwent N-dealkylation, oxidative deamination, and oxidative ring opening in humans.TPN729 has been reported as a novel phosphodiesterase type 5 (PDE5) inhibitor to treat erectile dysfunction, and is currently being tested in clinical trials. In addition to the potent inhibition against PDE5, TPN729 is regarded as a better alternative to provide fewer side effects and better patient compliance. Given the potential therapeutic benefits of TPN729, it is of great importance to elucidate its metabolic characteristics in drug development. This study is the first to investigate the metabolic fate of TPN729 in humans. A rapid and reliable analytical method based on ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometry (UPLC/Q–TOF MS) was established to investigate the metabolic profiles of TPN729 in human plasma, urine, and feces after its oral administration. As a result, a total of 22 metabolites were identified, of which seven were confirmed in comparison with the reference substances. The incubations of the metabolite references in human hepatocytes and aldehyde trapping experiment were further conducted to investigate its metabolic pathways. The results of the present study indicated the extensive metabolism of TPN729 in humans, including oxidative deamination, oxidative ring opening, N-dealkylation, N-oxidation, hydroxylation, dehydrogenation, lactam formation, and glucuronidation. M3 resulting from N-dealkylation was the major circulating substance detected in human plasma. The principal metabolites detected in human feces were products of oxidative deamination and oxidative ring opening. The parent drug was identified as the major component in urine. Taken together, this study provided valuable information on the metabolic fate of TPN729 in humans, and applicable analytical strategies for rapid metabolic elucidation in complex matrix samples through the useful and reliable UPLC/Q–TOF MS technique.
Co-reporter:Yunting Zhu, Liang Li, Ge Zhang, Hong Wan, Changyong Yang, Xingxing Diao, Xiaoyan Chen, Lianshan Zhang, Dafang Zhong
Journal of Chromatography B 2016 Volumes 1033–1034() pp:117-127
Publication Date(Web):15 October 2016
DOI:10.1016/j.jchromb.2016.08.009
•A novel strategy using UPLC/Q-TOF MS was established to characterize pyrotinib metabolites in humans.•The metabolic characteristics of pyrotinib in humans were first reported.•16 phase I and 8 phase II metabolites were detected and identified.•The structures of the major metabolites detected in humans were confirmed by comparison with the reference standards.•The primary enzymes responsible for the biotransformation of pyrotinib were identified.Pyrotinib is a novel irreversible tyrosine kinase inhibitor developed for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer. The results of phase I clinical trial demonstrated that pyrotinib was well tolerated and exhibited potent antitumor activity. As a promising therapeutic agent for HER2-positive breast cancer, it is of great importance to investigate the biotransformation of pyrotinib in humans and identify the major enzymes involved in its metabolism during its early stage of development for safety consideration. For this purpose, a robust analytical method based on ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometry (UPLC/Q-TOF MS) was established to characterize the metabolites of pyrotinib in human plasma, feces, and urine, and identify the primary enzymes responsible for its metabolism. As a result, a total of 24 metabolites were identified, including 16 phase I metabolites resulting from dealkylation, oxidation, dehydrogenation, and carbonylation, and 8 phase II metabolites originating from cysteine and N-acetylcysteine conjugation. Pyrotinib was absorbed into blood by 1 h, reached its peak level at 4 h, and afterwards underwent slow elimination. The principal metabolites detected in humans (M1, M2, and M5) were products resulting from O-depicoline and pyrrolidine lactam formation, whose structures have been confirmed by the synthetic references. In addition, fecal clearance was the major route of excretion for pyrotinib. Further phenotyping experiment proved that CYP3A4 was the most active enzyme responsible for the biotransformation of pyrotinib, implying the vital necessity of the assessment of the potential CYP3A-mediated drug–drug interactions in humans. Taken together, this study provided valuable metabolic data to explicate the dynamic process of pyrotinib in humans, and important reference basis for its safety evaluation and rational clinical application. The results will also benefit the assessment of the contributions to the overall activity or toxicity from the key metabolites.
Co-reporter:Jiangbo Du, Zhiyu Ma, Yifan Zhang, Ting Wang, Xiaoyan Chen, Dafang Zhong
Journal of Pharmaceutical and Biomedical Analysis 2013 Volume 86() pp:182-188
Publication Date(Web):December 2013
DOI:10.1016/j.jpba.2013.07.048
•The first chiral LC–MS/MS method for ornidazole quantification in human plasma.•Separation was performed on a Chiral-AGP column operated in reverse-phase mode.•Baseline resolution within 7.5 min leads to a huge reduction of total run time.•Deuterium labeled ISs were used to compensate for matrix effects.•Sensitivity was highly improved with smaller amount of plasma.A rapid, sensitive, and enantioselective method was developed and validated for determination of ornidazole enantiomers in human plasma by liquid chromatography–tandem mass spectrometry. Ornidazole enantiomers were extracted from 100 μl of plasma using ethyl acetate. Baseline chiral separation (Rs = 2.0) was obtained within 7.5 min on a Chiral-AGP column (150 mm × 4.0 mm, 5 μm) using an isocratic mobile phase of 10 mM ammonium acetate/acetic acid (100/0.01, v/v). Stable isotopically labeled R-(+)-d5-ornidazole and S-(−)-d5-ornidazole were synthesized as internal standards. Acquisition of mass spectrometric data was performed in multiple reaction monitoring mode via positive electrospray ionization, using the transitions of m/z 220 → 128 for ornidazole enantiomers, and m/z 225 → 128 for d5-ornidazole enantiomers. The method was linear in the concentration range of 0.030–10.0 μg/ml for each enantiomer. The lower limit of quantification for each enantiomer was 0.030 μg/ml. The relative standard deviation values of intra- and inter-day precision were 1.8–6.2% and 1.5–10.2% for R-(+)-ornidazole and S-(−)-ornidazole, respectively. The relative error values of accuracy ranged from −4.5% to 1.2% for R-(+)-ornidazole and from −5.4% to −0.8% for S-(−)-ornidazole. The validated method was successfully applied to a stereoselective pharmacokinetic study of ornidazole after oral administration of 1000 mg racemic ornidazole.Representative enantioselective MRM chromatograms for R-(+)-ornidazole (I), S-(−)-ornidazole (II), R-(+)-d5-ornidazole (IS; III), and S-(−)-d5-ornidazole (IS; IV) in human plasma. (A) Blank plasma sample. (B) Blank plasma sample spiked with R-(+)-ornidazole (0.030 μg/ml), S-(−)-ornidazole (0.030 μg/ml), R-(+)-d5-ornidazole (1.00 μg/ml), and S-(−)-d5-ornidazole (1.00 μg/ml). (C) Plasma sample at 1.5 h after oral administration of 1000 mg racemic ornidazole to a volunteer.
Co-reporter:Lishan Lin, Zhiwei Gao, Xiaoyan Chen, Dafang Zhong
Journal of Pharmaceutical and Biomedical Analysis 2013 Volume 86() pp:49-55
Publication Date(Web):December 2013
DOI:10.1016/j.jpba.2013.07.003
•Simultaneously determine allitinib and its two major metabolites in human plasma.•Allitinib and its metabolites have different physicochemical properties.•A simple protein precipitation and short chromatographic run time were achieved.•The method shows advantages of high selectivity and reproducibility.•The method was successfully applied to clinical study of allitinib tosylate.Allitinib, also known as AST1306, is a novel irreversible inhibitor of the epidermal growth factor receptors 1 and 2. Allitinib is currently used in clinical trial to treat solid tumors. A previous study showed that allitinib is extensively metabolized in humans. Amide hydrolysis metabolite (M6) and 29,30-dihydrodiol allitinib (M10) are the major metabolites in circulation. To study the pharmacokinetics of allitinib and its two major metabolites in cancer patients, a rapid, sensitive and reliable LC–MS/MS method was developed and validated for the simultaneous determination of allitinib, M6 and M10 in human plasma. After simple protein precipitation, the analytes and the combined internal standards (lapatinib and NB-2, an analog of allitinib) were separated on a Zorbax Eclipase XDB C18 column (50 mm × 4.6 mm, 1.8 μm, Agilent) using a mobile phase of 5 mM ammonium acetate with 0.1% formic acid (phase A) and 50% (v/v) methanol in acetonitrile (phase B) with gradient elution. Mass spectrometric detection was conducted by atmospheric-pressure chemical ionization in positive ion multiple reaction monitoring modes using AB Sciex Triple Quad 6500 system. Linear calibration curves were obtained for the following concentration range: 0.300−200 ng/ml for allitinib; 0.030−20.0 ng/ml for M6; and 0.075−50.0 ng/ml for M10. Intra-day and inter-day accuracy and precision were within the acceptable limits of ±15% at all of the concentrations. The method was successfully applied to a preliminary clinical pharmacokinetic study following oral administration of allitinib tosylate tablets in cancer patients.Product ion mass spectra of [M+H]+ ion of (A) allitinib, (B) M6, (C) M10, (D) NB-2, and (E) lapatinib.

![3-Pyridinecarboxamide, N-[4-[(2-amino-3-chloro-4-pyridinyl)oxy]-3-fluorophenyl]-5-(4-fluorophenyl)-1,4-dihydro-4-oxo-](/data/chemimg/390900/1174046-72-0.png)
![3-Pyridinecarboxamide, N-[4-[(2-amino-3-chloro-4-pyridinyl)oxy]-3-fluorophenyl]-5-(4-fluorophenyl)-1,4-dihydro-4-oxo-](/data/chemimg/390900/1174046-72-0_b.png)
![Benzonitrile, 3-[1,6-dihydro-1-[[3-[5-[(1-methyl-4-piperidinyl)methoxy]-2-pyrimidinyl]phenyl]methyl]-6-oxo-3-pyridazinyl]-](/data/chemimg/345600/1100598-32-0.png)
![Benzonitrile, 3-[1,6-dihydro-1-[[3-[5-[(1-methyl-4-piperidinyl)methoxy]-2-pyrimidinyl]phenyl]methyl]-6-oxo-3-pyridazinyl]-](/data/chemimg/345600/1100598-32-0_b.png)
![Benzamide, 2-fluoro-N-methyl-4-[7-(6-quinolinylmethyl)imidazo[1,2-b][1,2,4]triazin-2-yl]-](/data/chemimg/315100/1029712-80-8.png)
![Benzamide, 2-fluoro-N-methyl-4-[7-(6-quinolinylmethyl)imidazo[1,2-b][1,2,4]triazin-2-yl]-](/data/chemimg/315100/1029712-80-8_b.png)
![2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyr azol-1-yl)ethanol](http://img.cochemist.com/ccimg/957000/956905-27-4.png)
![2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyr azol-1-yl)ethanol](http://img.cochemist.com/ccimg/957000/956905-27-4_b.png)
![1,1-Cyclopropanedicarboxamide, N-[2-fluoro-4-[[2-[[[4-(4-methyl-1-piperazinyl)-1-piperidinyl]carbonyl]amino]-4-pyridinyl]oxy]phenyl]-N'-(4-fluorophenyl)-](/data/chemimg/2243600/928037-13-2.png)
![1,1-Cyclopropanedicarboxamide, N-[2-fluoro-4-[[2-[[[4-(4-methyl-1-piperazinyl)-1-piperidinyl]carbonyl]amino]-4-pyridinyl]oxy]phenyl]-N'-(4-fluorophenyl)-](/data/chemimg/2243600/928037-13-2_b.png)
![1H-Pyrazole-4-carboxamide, 2,3-dihydro-1-(2-hydroxy-2-methylpropyl)-N-[5-[(7-methoxy-4-quinolinyl)oxy]-2-pyridinyl]-5-methyl-3-oxo-2-phenyl-](/data/chemimg/1589500/913376-83-7.png)
![1H-Pyrazole-4-carboxamide, 2,3-dihydro-1-(2-hydroxy-2-methylpropyl)-N-[5-[(7-methoxy-4-quinolinyl)oxy]-2-pyridinyl]-5-methyl-3-oxo-2-phenyl-](/data/chemimg/1589500/913376-83-7_b.png)


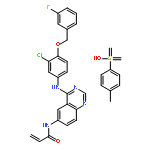
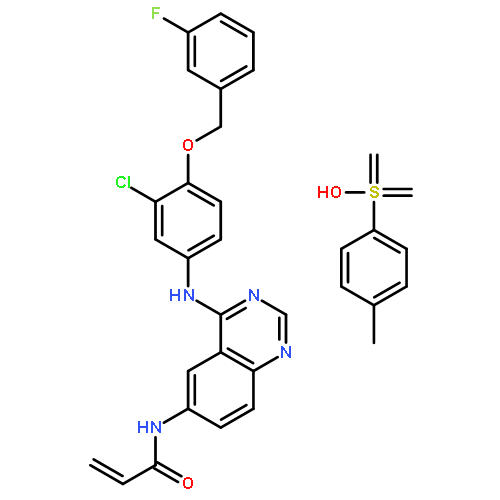


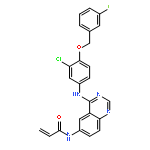
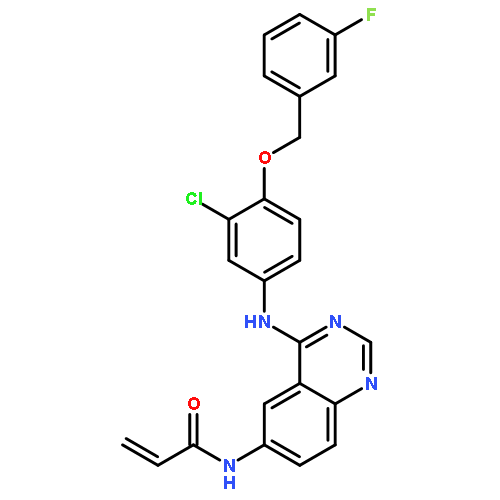
![Benzeneacetamide, N-[[[3-fluoro-4-[[2-(1-methyl-1H-imidazol-4-yl)thieno[3,2-b]pyridin-7-yl]oxy]phenyl]amino]thioxomethyl]-](/data/chemimg/1587400/875337-44-3.png)
![Benzeneacetamide, N-[[[3-fluoro-4-[[2-(1-methyl-1H-imidazol-4-yl)thieno[3,2-b]pyridin-7-yl]oxy]phenyl]amino]thioxomethyl]-](/data/chemimg/1587400/875337-44-3_b.png)





















![Benzenesulfonamide,3-(4,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-](http://img.cochemist.com/ccimg/319500/319491-68-4.png)
![Benzenesulfonamide,3-(4,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-](http://img.cochemist.com/ccimg/319500/319491-68-4_b.png)
![calcium,(3S,5S)-7-[2-(4-fluorophenyl)-4-[(2-hydroxyphenyl)carbamoyl]-3-phenyl-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoate](http://img.cochemist.com/ccimg/266000/265989-46-6.png)
![calcium,(3S,5S)-7-[2-(4-fluorophenyl)-4-[(2-hydroxyphenyl)carbamoyl]-3-phenyl-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoate](http://img.cochemist.com/ccimg/266000/265989-46-6_b.png)
![(1-Hydroxy-2-(imidazo[1,2-a]pyridin-3-yl)ethane-1,1-diyl)diphosphonic acid](http://img.cochemist.com/ccimg/180100/180064-38-4.png)
![(1-Hydroxy-2-(imidazo[1,2-a]pyridin-3-yl)ethane-1,1-diyl)diphosphonic acid](http://img.cochemist.com/ccimg/180100/180064-38-4_b.png)

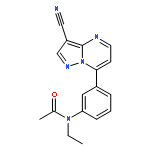
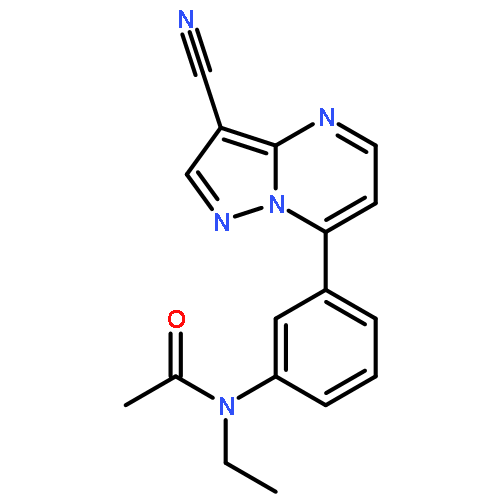

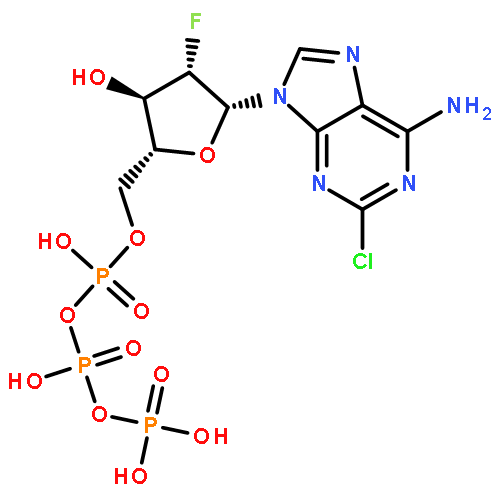


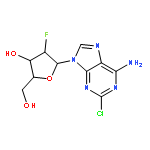
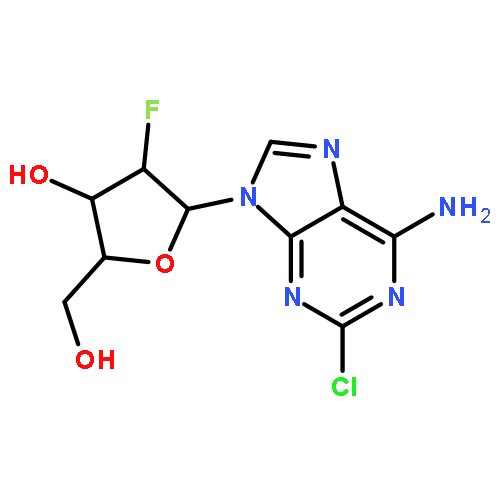


![Benzonitrile, 4-[(6,7-dihydro-7-hydroxy-5H-cyclopentapyrimidin-4-yl)amino]-](/data/chemimg/315300/105365-76-2.png)
![Benzonitrile, 4-[(6,7-dihydro-7-hydroxy-5H-cyclopentapyrimidin-4-yl)amino]-](/data/chemimg/315300/105365-76-2_b.png)

![Thieno[3,2-c]pyridine-5(4H)-aceticacid, a-(2-chlorophenyl)-6,7-dihydro-,methyl ester](http://img.cochemist.com/ccimg/90100/90055-48-4.png)
![Thieno[3,2-c]pyridine-5(4H)-aceticacid, a-(2-chlorophenyl)-6,7-dihydro-,methyl ester](http://img.cochemist.com/ccimg/90100/90055-48-4_b.png)
![N-[2-(dimethylamino)ethyl]acridine-4-carboxamide](http://img.cochemist.com/ccimg/89500/89459-25-6.png)
![N-[2-(dimethylamino)ethyl]acridine-4-carboxamide](http://img.cochemist.com/ccimg/89500/89459-25-6_b.png)


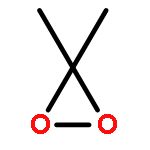

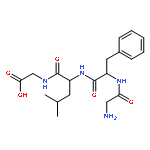
![L-gamma-glutamyl-S-[(1R)-7-(hydroxymethyl)-2,3-dihydro-1H-pyrrolizin-1-yl]-L-cysteinylglycine](http://img.cochemist.com/ccimg/64700/64660-86-2.png)
![L-gamma-glutamyl-S-[(1R)-7-(hydroxymethyl)-2,3-dihydro-1H-pyrrolizin-1-yl]-L-cysteinylglycine](http://img.cochemist.com/ccimg/64700/64660-86-2_b.png)


![5-(2-Chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/55200/55142-85-3.png)
![5-(2-Chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/55200/55142-85-3_b.png)


![Octadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/51100/51063-97-9.png)
![Octadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/51100/51063-97-9_b.png)
![Hexadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/40300/40290-32-2.png)
![Hexadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/40300/40290-32-2_b.png)

![Hexadecanoic acid,1,1'-[(1S)-1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/30400/30334-71-5.png)
![Hexadecanoic acid,1,1'-[(1S)-1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/30400/30334-71-5_b.png)


![Octadecanoic acid, 3-hydroxy-2-[(1-oxohexadecyl)oxy]propyl ester](http://img.cochemist.com/ccimg/14100/14015-55-5.png)
![Octadecanoic acid, 3-hydroxy-2-[(1-oxohexadecyl)oxy]propyl ester](http://img.cochemist.com/ccimg/14100/14015-55-5_b.png)





![9,12-Octadecadienoicacid (9Z,12Z)-, 1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/2500/2442-62-8.png)
![9,12-Octadecadienoicacid (9Z,12Z)-, 1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/2500/2442-62-8_b.png)
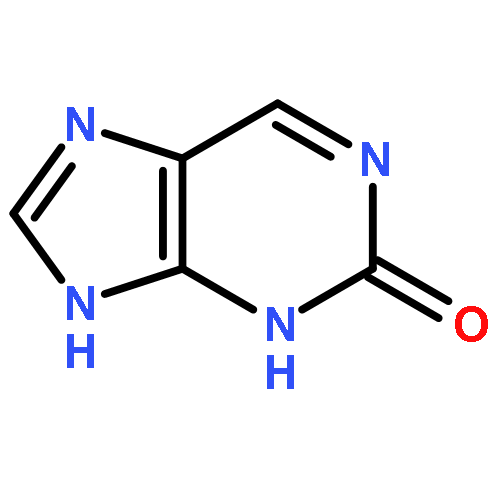




![Glycine, N-[(3a,5b,7a)-3,7-dihydroxy-24-oxocholan-24-yl]-](http://img.cochemist.com/ccimg/700/640-79-9.png)
![Glycine, N-[(3a,5b,7a)-3,7-dihydroxy-24-oxocholan-24-yl]-](http://img.cochemist.com/ccimg/700/640-79-9_b.png)



![5'-O-[hydroxy(sulfooxy)phosphoryl]adenosine 3'-(dihydrogen phosphate)](http://img.cochemist.com/ccimg/500/482-67-7.png)
![5'-O-[hydroxy(sulfooxy)phosphoryl]adenosine 3'-(dihydrogen phosphate)](http://img.cochemist.com/ccimg/500/482-67-7_b.png)


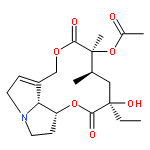
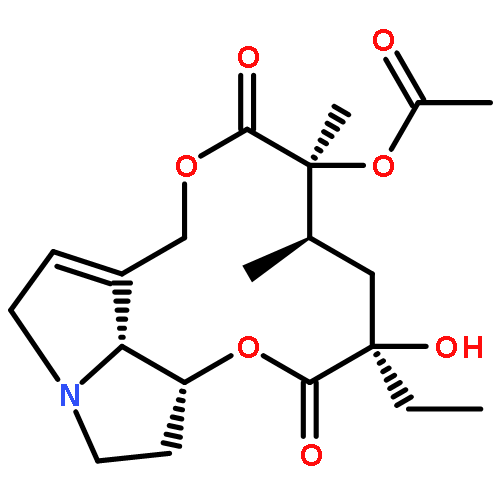
![[1,6]Dioxacyclododecino[2,3,4-gh]pyrrolizine-2,7-dione,3-ethylidene-3,4,5,6,9,11,13,14,14a,14b-decahydro-6-hydroxy-6-(hydroxymethyl)-5-methyl-,(3Z,5R,6S,14aR,14bR)-](http://img.cochemist.com/ccimg/500/480-54-6.png)
![[1,6]Dioxacyclododecino[2,3,4-gh]pyrrolizine-2,7-dione,3-ethylidene-3,4,5,6,9,11,13,14,14a,14b-decahydro-6-hydroxy-6-(hydroxymethyl)-5-methyl-,(3Z,5R,6S,14aR,14bR)-](http://img.cochemist.com/ccimg/500/480-54-6_b.png)
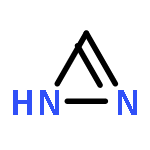
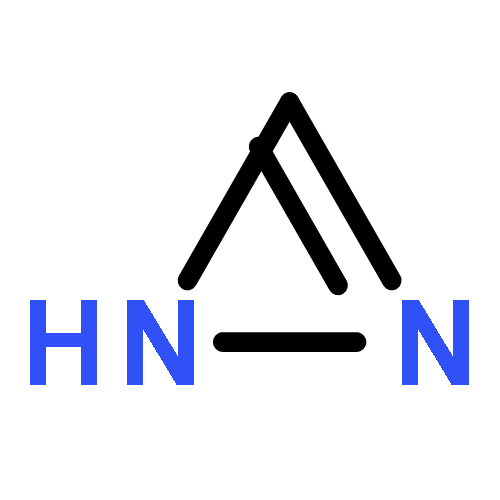






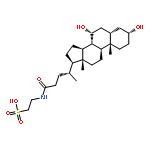
![2-[[(4r)-4-[(3r,5s,7r,8r,9s,10s,12s,13r,14s,17r)-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]ethanesulfonic Acid](http://img.cochemist.com/ccimg/100/81-24-3.png)
![2-[[(4r)-4-[(3r,5s,7r,8r,9s,10s,12s,13r,14s,17r)-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]ethanesulfonic Acid](http://img.cochemist.com/ccimg/100/81-24-3_b.png)












![Butanedioic acid,1-[4-[[1-[[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]thio]-1-methylethyl]thio]-2,6-bis(1,1-dimethylethyl)phenyl]ester](http://img.cochemist.com/ccimg/216200/216167-82-7.png)
![Butanedioic acid,1-[4-[[1-[[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]thio]-1-methylethyl]thio]-2,6-bis(1,1-dimethylethyl)phenyl]ester](http://img.cochemist.com/ccimg/216200/216167-82-7_b.png)





