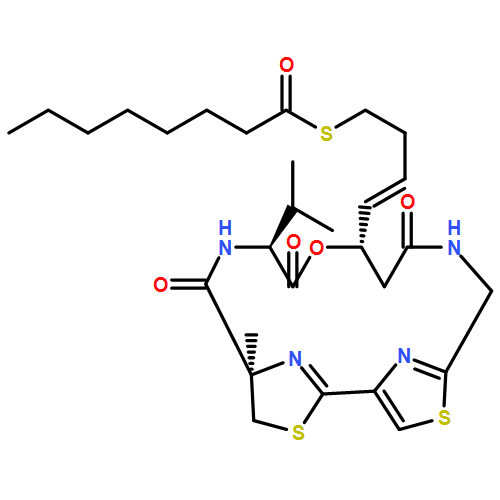
Co-reporter:Weijing Cai, Qi-Yin Chen, Long H. Dang, and Hendrik Luesch
ACS Medicinal Chemistry Letters October 12, 2017 Volume 8(Issue 10) pp:1007-1007
Publication Date(Web):September 18, 2017
DOI:10.1021/acsmedchemlett.7b00192
Renal, hepatocellular, and neuroendocrine carcinomas are known as highly vascularized tumors. Although vascular endothelial growth factor A (VEGF-A)-targeted therapies have shown efficacy in the treatment of these cancers, drug resistance is a major concern and might be mediated by interleukin 6 (IL-6). Furthermore, upon antiangiogenic drug exposure, tumor cells may adapt to survive in a vascular-independent manner. Apratoxins are potent marine-derived cytotoxic in vivo-active agents, preventing cotranslational translocation in the secretory pathway, and show promise to overcome resistance by targeting angiogenesis and tumor growth simultaneously. We designed and synthesized a novel apratoxin analogue, apratoxin S10, with a balanced potency and stability as well as synthetic accessibility and scalability. We showed that apratoxin S10 potently inhibits both angiogenesis in vitro and growth of cancer cells from vascularized tumors. Apratoxin S10 down-regulated vascular endothelial growth factor receptor 2 (VEGFR2) on endothelial cells and blocked the secretion of VEGF-A and IL-6 from cancer cells. It inhibited cancer cell growth through down-regulation of multiple receptor tyrosine kinases (RTKs) and compares favorably to currently approved RTK inhibitors in both angiogenesis and cancer cell growth.Keywords: Antiangiogenic agents; antiproliferative agents; cotranslational translocation inhibitor; receptor tyrosine kinases; total synthesis; vascular endothelial growth factor;
Co-reporter:Fatma H. Al-Awadhi, Brian K. Law, Valerie J. Paul, and Hendrik Luesch
Journal of Natural Products November 22, 2017 Volume 80(Issue 11) pp:2969-2969
Publication Date(Web):October 31, 2017
DOI:10.1021/acs.jnatprod.7b00551
Three new modified peptides named grassystatins D–F (1–3) were discovered from a marine cyanobacterium from Guam. Their structures were elucidated using NMR spectroscopy and mass spectrometry. The hallmark structural feature in the peptides is a statine unit, which contributes to their aspartic protease inhibitory activity preferentially targeting cathepsins D and E. Grassystatin F (3) was the most potent analogue, with IC50 values of 50 and 0.5 nM against cathepsins D and E, respectively. The acidic tumor microenvironment is known to increase the activation of some of the lysosomal proteases associated with tumor metastasis such as cathepsins. Because cathepsin D is a biomarker in aggressive forms of breast cancer and linked to poor prognosis, the effects of cathepsin D inhibition by 1 and 3 on the downstream cellular substrates cystatin C and PAI-1 were investigated. Furthermore, the functional relevance of targeting cathepsin D substrates was evaluated by examining the effect of 1 and 3 on the migration of MDA-MD-231 cells. Grassystatin F (3) inhibited the cleavage of cystatin C and PAI-1, the activities of their downstream targets cysteine cathepsins and tPA, and the migration of the highly aggressive triple negative breast cancer cells, phenocopying the effect of siRNA-mediated knockdown of cathepsin D.
Co-reporter:Hendrik Luesch
Biochemical Pharmacology 2017 Volume 139(Volume 139) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.bcp.2017.06.054
Natural products from marine cyanobacteria cover therapeutically relevant chemical space. This is underscored by the approval of brentuximab vedotin, an antibody-drug conjugate with a cytotoxic payload that is derived from dolastatin 10 which is produced by the marine cyanobacterium Caldora penicillata. In addition to dolastatin 10, we have discovered novel cytotoxins with different mechanisms of action, including apratoxins. We characterized the targets and mechanisms of action of prioritized cytotoxins to identify biomarkers for preclinical studies, performed structure–activity relationship studies to modulate activity and selectivity profiles, and investigated their efficacy using tumor xenograft models to assess their potential as anticancer drug leads.
Co-reporter:Bumki Kim, Ranjala Ratnayake, Hyunji Lee, Guqin Shi, Sabrina L. Zeller, Chenglong Li, Hendrik Luesch, Jiyong Hong
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 12(Issue 12) pp:
Publication Date(Web):15 June 2017
DOI:10.1016/j.bmc.2017.03.071
Histone acetylation is an extensively investigated post-translational modification that plays an important role as an epigenetic regulator. It is controlled by histone acetyl transferases (HATs) and histone deacetylases (HDACs). The overexpression of HDACs and consequent hypoacetylation of histones have been observed in a variety of different diseases, leading to a recent focus of HDACs as attractive drug targets. The natural product largazole is one of the most potent natural HDAC inhibitors discovered so far and a number of largazole analogs have been prepared to define structural requirements for its HDAC inhibitory activity. However, previous structure–activity relationship studies have heavily investigated the macrocycle region of largazole, while there have been only limited efforts to probe the effect of various zinc-binding groups (ZBGs) on HDAC inhibition. Herein, we prepared a series of largazole analogs with various ZBGs and evaluated their HDAC inhibition and cytotoxicity. While none of the analogs tested were as potent or selective as largazole, the Zn2+-binding affinity of each ZBG correlated with HDAC inhibition and cytotoxicity. We expect that our findings will aid in building a deeper understanding of the role of ZBGs in HDAC inhibition as well as provide an important basis for the future development of new largazole analogs with non-thiol ZBGs as novel therapeutics for cancer.The overexpression of HDACs and consequent hypoacetylation of histones have been observed in a variety of different diseases, leading to a recent focus of HDACs as attractive drug targets. The natural product largazole is one of the most potent natural HDAC inhibitors discovered so far. To probe the effect of various zinc-binding groups (ZBGs) on HDAC inhibition. We prepared a series of largazole analogs with various ZBGs and evaluated their HDAC inhibition and cytotoxicity.Download high-res image (151KB)Download full-size image
Co-reporter:Ping Wu, Weijing Cai, Qi-Yin Chen, Senhan Xu, Ruwen Yin, Yingxia Li, Wei Zhang, and Hendrik Luesch
Organic Letters 2016 Volume 18(Issue 20) pp:5400-5403
Publication Date(Web):October 10, 2016
DOI:10.1021/acs.orglett.6b02780
Apratoxin E provided the inspiration for the design of apratoxin A/E hybrids under preclinical development. Through total synthesis using two different strategies, it was determined that the originally proposed configuration of the thiazoline at C30 is opposite from that in apratoxin A, in contrast to previous assumptions on biosynthetic grounds. The epimer and true natural apratoxin E were synthesized, and the biological activities were evaluated.
Co-reporter:Michelle S. Bousquet, Jia Jia Ma, Ranjala Ratnayake, Pamela A. Havre, Jin Yao, Nam H. Dang, Valerie J. Paul, Thomas J. Carney, Long H. Dang, and Hendrik Luesch
ACS Chemical Biology 2016 Volume 11(Issue 5) pp:1322
Publication Date(Web):March 3, 2016
DOI:10.1021/acschembio.5b00860
Colorectal cancer (CRC) is a genetic disease, due to progressive accumulation of mutations in oncogenes and tumor suppressor genes. Large scale genomic sequencing projects revealed >100 mutations in any individual CRC. Many of these mutations are likely passenger mutations, and fewer are driver mutations. Of these, activating mutations in RAS proteins are essential for cancer initiation, progression, and/or resistance to therapy. There has been significant interest in developing drugs targeting mutated cancer gene products or downstream signaling pathways. Due to the number of mutations involved and inherent redundancy in intracellular signaling, drugs targeting one mutation or pathway have been either ineffective or led to rapid resistance. We have devised a strategy whereby multiple cancer pathways may be simultaneously targeted for drug discovery. For proof-of-concept, we targeted the oncogenic KRAS and HIF pathways, since oncogenic KRAS has been shown to be required for cancer initiation and progression, and HIF-1α and HIF-2α are induced by the majority of mutated oncogenes and tumor suppressor genes in CRC. We have generated isogenic cell lines defective in either oncogenic KRAS or both HIF-1α and HIF-2α and subjected them to multiplex genomic, siRNA, and high-throughput small molecule screening. We have identified potential drug targets and compounds for preclinical and clinical development. Screening of our marine natural product library led to the rediscovery of the microtubule agent dolastatin 10 and the class I histone deacetylase (HDAC) inhibitor largazole to inhibit oncogenic KRAS and HIF pathways. Largazole was further validated as an antiangiogenic agent in a HIF-dependent manner in human cells and in vivo in zebrafish using a genetic model with activated HIF. Our general strategy, coupling functional genomics with drug susceptibility or chemical-genetic interaction screens, enables the identification of potential drug targets and candidates with requisite selectivity. Molecules prioritized in this manner can easily be validated in suitable zebrafish models due to the genetic tractability of the system. Our multidimensional platform with cellular and organismal components can be extended to larger scale multiplex screens that include other mutations and pathways.
Co-reporter:Sarath P. Gunasekera; Lorelie Imperial; Christiana Garst; Ranjala Ratnayake; Long H. Dang; Valerie J. Paul
Journal of Natural Products 2016 Volume 79(Issue 7) pp:1867-1871
Publication Date(Web):July 5, 2016
DOI:10.1021/acs.jnatprod.6b00203
The isolation, structure determination, and biological activities of a new linear pentapeptide, caldoramide (5), from the marine cyanobacterium Caldora penicillata from Florida are described. Caldoramide (5) has structural similarities to belamide A (4), dolastatin 10 (1), and dolastatin 15 (2). We profiled caldoramide against parental HCT116 colorectal cancer cells and isogenic cells lacking oncogenic KRAS or hypoxia-inducible factors 1α (HIF-1α) and 2α (HIF-2α). Caldoramide (5) showed differential cytotoxicity for cells containing both oncogenic KRAS and HIF over the corresponding knockout cells. LCMS dereplication indicated the presence of caldoramide (5) in a subset of C. penicillata samples.
Co-reporter:Fatma H. Al-Awadhi, Ranjala Ratnayake, Valerie J. Paul, Hendrik Luesch
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 15) pp:3276-3282
Publication Date(Web):1 August 2016
DOI:10.1016/j.bmc.2016.04.062
In search of novel protease inhibitors with therapeutic potential, our efforts exploring the marine cyanobacterium Lyngbya sp. have led to the discovery of tasiamide F (1), which is an analogue of tasiamide B (2). The structure was elucidated using a combination of NMR spectroscopy and mass spectrometry. The key structural feature in 1 is the presence of the Phe-derived statine core, which contributes to its aspartic protease inhibitory activity. The antiproteolytic activity of 1 and 2 was evaluated in vitro against cathepsins D and E, and BACE1. Tasiamide F (1) displayed IC50 values of 57 nM, 23 nM, and 0.69 μM, respectively, indicating greater selectivity for cathepsins over BACE1 compared with tasiamide B (2). Molecular docking experiments were carried out for compounds 1 and 2 against cathepsins D and E to rationalize their activity towards these proteases. The dysregulated activities of cathepsins D and E have been implicated in cancer and modulation of immune responses, respectively, and these proteases represent potential therapeutic targets.
Co-reporter:Danmeng Luo, Qi-Yin Chen, and Hendrik Luesch
The Journal of Organic Chemistry 2016 Volume 81(Issue 2) pp:532-544
Publication Date(Web):December 28, 2015
DOI:10.1021/acs.joc.5b02386
Lyngbyastatin 7 (1) is a marine cyanobacteria-derived lariat-type cyclic depsipeptide of which the macrocyclic core possesses modified amino acids, including a featured 3-amino-6-hydroxy-2-piperidone (Ahp) moiety and a (Z)-2-amino-2-butenoic acid (Abu) moiety. The first total synthesis of 1 was successfully established via 31 steps, and the conditions of several crucial steps were optimized to ensure smooth operations. The previously reported structural assignment and elastase inhibitory activity of the isolated natural product were confirmed. According to the extensive in vitro biological evaluation, compound 1 displayed low nanomolar IC50 in blocking elastase activity and strong ability in protecting bronchial epithelial cells against elastase-induced antiproliferation and abrogating the elastase-triggered induction of pro-inflammatory cytokine expression. Its overall performance was superior over sivelestat, the only approved small molecule drug targeting elastase, which indicated its potential in developing as a pharmacotherapeutic against elastase-mediated pathologies. The success in total synthesis, designed with a novel convergent strategy, not only overcame the supply issue for thorough preclinical studies but also paved the way for convenient synthesis of analogues with improved potency and druglike properties.
Co-reporter:Lilibeth A. Salvador-Reyes and Hendrik Luesch
Natural Product Reports 2015 vol. 32(Issue 3) pp:478-503
Publication Date(Web):09 Jan 2015
DOI:10.1039/C4NP00104D
Covering: up to 2014
Marine cyanobacteria are an ancient group of organisms and prolific producers of bioactive secondary metabolites. These compounds are presumably optimized by evolution over billions of years to exert high affinity for their intended biological target in the ecologically relevant organism but likely also possess activity in different biological contexts such as human cells. Screening of marine cyanobacterial extracts for bioactive natural products has largely focused on cancer cell viability; however, diversification of the screening platform led to the characterization of many new bioactive compounds. Targets of compounds have oftentimes been elusive if the compounds were discovered through phenotypic assays. Over the past few years, technology has advanced to determine mechanism of action (MOA) and targets through reverse chemical genetic and proteomic approaches, which has been applied to certain cyanobacterial compounds and will be discussed in this review. Some cyanobacterial molecules are the most-potent-in-class inhibitors and therefore may become valuable tools for chemical biology to probe protein function but also be templates for novel drugs, assuming in vitro potency translates into cellular and in vivo activity. Our review will focus on compounds for which the direct targets have been deciphered or which were found to target a novel pathway, and link them to disease states where target modulation may be beneficial.
Co-reporter:Danmeng Luo, Hendrik Luesch
Chemistry & Biology 2015 Volume 22(Issue 5) pp:565-567
Publication Date(Web):21 May 2015
DOI:10.1016/j.chembiol.2015.05.003
In this issue of Chemistry & Biology, Arita et al. (2015) report that theonellamides can specifically recognize cholesterol in liquid-disordered environment, modulate membrane order, and change cell morphology and thus may serve as probes to unveil the enigmatic nature of cell membranes.
Co-reporter:Lilibeth A. Salvador-Reyes, Niclas Engene, Valerie J. Paul, and Hendrik Luesch
Journal of Natural Products 2015 Volume 78(Issue 3) pp:486-492
Publication Date(Web):January 30, 2015
DOI:10.1021/np500931q
Combined phylogenetic and HPLC-MS-based natural products dereplication methods aimed at identifying cyanobacterial collections containing the potent cytotoxins largazole, dolastatin 10, and symplostatin 1 were developed. The profiling of the phylogeny, chemical space, and antiproliferative activity of cyanobacterial collections served to streamline the prioritization of samples for the discovery of new secondary metabolites. The dereplication methods highlighted the biosynthetic potential and combinatorial pharmacology employed by marine cyanobacteria. We found that largazole was always coproduced with dolastatin 10 or with symplostatin 1 and consequently tested combinations of these agents against colon cancer cells. Combinatorial regimens of largazole and dolastatin 10 aimed at curbing the growth of HCT116 cancer cells showed cooperative activity.
Co-reporter:Lilibeth A. Salvador-Reyes, Jennifer Sneed, Valerie J. Paul, and Hendrik Luesch
Journal of Natural Products 2015 Volume 78(Issue 8) pp:1957-1962
Publication Date(Web):July 23, 2015
DOI:10.1021/acs.jnatprod.5b00293
Cytotoxicity-guided fractionation of a Guamanian cyanobacterial collection yielded the new compounds amantelides A (1) and B (2). These polyketides are characterized by a 40-membered macrolactone ring consisting of a 1,3-diol and contiguous 1,5-diol units and a tert-butyl substituent. Amantelide A (1) displayed potent cytotoxicity with submicromolar IC50 against HT29 colorectal adenocarcinoma and HeLa cervical carcinoma cell lines. Acetylation of the hydroxy group at C-33 in 2 caused a close to 10-fold decrease in potency. Exhaustive acetylation of the hydroxy groups abrogated the antiproliferative activity of amantelide A (1) by 20–67-fold. Further bioactivity assessment of 1 against bacterial pathogens and marine fungi indicated a broad spectrum of bioactivity.
Co-reporter:Qi-Yin Chen ; Yanxia Liu ; Weijing Cai
Journal of Medicinal Chemistry 2014 Volume 57(Issue 7) pp:3011-3029
Publication Date(Web):March 24, 2014
DOI:10.1021/jm4019965
Apratoxins are cytotoxic natural products originally isolated from marine cyanobacteria that act by preventing cotranslational translocation early in the secretory pathway to downregulate receptor levels and inhibit growth factor secretion, leading to potent antiproliferative activity. Through rational design and total synthesis of an apratoxin A/E hybrid, apratoxin S4 (1a), we have previously improved the antitumor activity and tolerability in vivo. Compound 1a and newly designed analogues apratoxins S7–S9 (1b–d), with various degrees of methylation at C34 (1b,c) or epimeric configuration at C30 (1d), were efficiently synthesized utilizing improved procedures. Optimizations have been applied to the synthesis of key intermediate aldehyde 7 and further include the application of Leighton’s silanes and modifications of Kelly’s methods to induce thiazoline ring formation in other crucial steps of the apratoxin synthesis. Apratoxin S9 (1d) exhibited increased activity with subnanomolar potency. Apratoxin S8 (1c) lacks the propensity to be deactivated by dehydration and showed efficacy in a human HCT116 xenograft mouse model.
Co-reporter:Lilibeth A. Salvador, Heekwang Park, Fatma H. Al-Awadhi, Yanxia Liu, Bumki Kim, Sabrina L. Zeller, Qi-Yin Chen, Jiyong Hong, and Hendrik Luesch
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 8) pp:905
Publication Date(Web):July 7, 2014
DOI:10.1021/ml500170r
Largazole is a potent and class I-selective histone deacetylase (HDAC) inhibitor purified from marine cyanobacteria and was demonstrated to possess antitumor activity. Largazole employs a unique prodrug strategy, via a thioester moiety, to liberate the bioactive species largazole thiol. Here we report alternate prodrug strategies to modulate the pharmacokinetic and pharmacodynamics profiles of new largazole-based compounds. The in vitro effects of largazole analogues on cancer cell proliferation and enzymatic activities of purified HDACs were comparable to the natural product. However, in vitro and in vivo histone hyperacetylation in HCT116 cells and implanted tumors, respectively, showed differences, particularly in the onset of action and oral bioavailability. These results indicate that, by employing a different approach to disguise the “warhead” moiety, the functional consequence of these prodrugs can be significantly modulated. Our data corroborate the role of the pharmacokinetic properties of this class of compounds to elicit the desired and timely functional response.Keywords: antitumor activity; histone deacetylases; Largazole; natural products; prodrugs
Co-reporter:Bumki Kim, Heekwang Park, Lilibeth A. Salvador, Patrick E. Serrano, Jason C. Kwan, Sabrina L. Zeller, Qi-Yin Chen, Soyoung Ryu, Yanxia Liu, Seongrim Byeon, Hendrik Luesch, Jiyong Hong
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 16) pp:3728-3731
Publication Date(Web):15 August 2014
DOI:10.1016/j.bmcl.2014.07.006
Largazole is a potent class I selective histone deacetylase (HDAC) inhibitor. The majority of largazole analogues to date have modified the thiazole–thiazoline and the warhead moiety. In order to elucidate class I-specific structure–activity relationships, a series of analogues with modifications in the valine or the linker region were prepared and evaluated for their class I isoform selectivity. The inhibition profile showed that the C2 position of largazole has an optimal steric requirement for efficient HDAC inhibition and that substitution of the trans-alkene in the linker with an aromatic group results in complete loss of activity. This data will aid the design of class I isoform selective HDAC inhibitors.
Co-reporter:Dr. Jason C. Kwan;Dr. Yanxia Liu;Dr. Ranjala Ratnayake;Ryo Hatano;Akiko Kuribara; Dr. Chiko Morimoto;Dr. Kei Ohnuma;Dr. Valerie J. Paul; Dr. Tao Ye; Dr. Hendrik Luesch
ChemBioChem 2014 Volume 15( Issue 6) pp:799-804
Publication Date(Web):
DOI:10.1002/cbic.201300762
Abstract
Natural products made by marine cyanobacteria are often highly modified peptides and depsipeptides that have the potential to act as inhibitors for proteases. In the interests of finding new protease inhibition activity and selectivity, grassypeptolide A (1) was screened against a panel of proteases and found to inhibit DPP8 selectively over DPP4. Grassypeptolides were also found to inhibit IL-2 production and proliferation in activated T-cells, consistent with a putative role of DPP8 in the immune system. These effects were also observed in Jurkat cells, and DPP activity in Jurkat cell cytosol was shown to be inhibited by grassypeptolides. In silico docking suggests two possible binding modes of grassypeptolides—at the active site of DPP8 and at one of the entrances to the internal cavity. Collectively these results suggest that grassypeptolides might be useful tool compounds in the study of DPP8 function.
Co-reporter:Rana Montaser, Valerie J. Paul, and Hendrik Luesch
Organic Letters 2013 Volume 15(Issue 16) pp:4050-4053
Publication Date(Web):August 5, 2013
DOI:10.1021/ol401396u
Novel bioactive lipids were identified from a Guamanian cyanobacterium, the Pseudomonas aeruginosa quorum sensing inhibitor pitinoic acid A (1) and the anti-inflammatory pitinoic acids B (2) and C. The structure of 2 was confirmed by synthesis, which also allowed for biological evaluation. Since 2 is an ester of pitinoic acids A and C, it represents a prodrug strategy to liberate dual biological activity for the management of P. aeruginosa infections and their associated inflammation.
Co-reporter:Lilibeth A. Salvador ; Kanchan Taori ; Jason S. Biggs ; Jean Jakoncic ; David A. Ostrov ; Valerie J. Paul
Journal of Medicinal Chemistry 2013 Volume 56(Issue 3) pp:1276-1290
Publication Date(Web):January 28, 2013
DOI:10.1021/jm3017305
We discovered new structural diversity to a prevalent, yet medicinally underappreciated, cyanobacterial protease inhibitor scaffold and undertook comprehensive protease profiling to reveal potent and selective elastase inhibition. Structure-activity relationship (SAR) studies and X-ray cocrystal structure analysis allowed a detailed assessment of critical and tunable structural elements. To realize the therapeutic potential of these cyclodepsipeptides, we probed the cellular effects of a novel and representative family member, symplostatin 5 (1), which attenuated the downstream cellular effects of elastase in an epithelial lung airway model system, alleviating clinical hallmarks of chronic pulmonary diseases such as cell death, cell detachment, and inflammation. This compound attenuated the effects of elastase on receptor activation, proteolytic processing of the adhesion protein ICAM-1, NF-κB activation, and transcriptomic changes, including the expression of pro-inflammatory cytokines IL1A, IL1B, and IL8. Compound 1 exhibited activity comparable to the clinically approved elastase inhibitor sivelestat in short-term assays and demonstrated superior sustained activity in longer-term assays.
Co-reporter:Rui Wang, Daniel E. Mason, Keith P. Choe, Alfred S. Lewin, Eric C. Peters, and Hendrik Luesch
ACS Chemical Biology 2013 Volume 8(Issue 8) pp:1764
Publication Date(Web):June 17, 2013
DOI:10.1021/cb4000103
The cell utilizes the Keap1/Nrf2-ARE signaling pathway to detoxify harmful chemicals in order to protect itself from oxidative stress and to maintain its reducing environment. When exposed to oxidative stress and xenobiotic inducers, the redox sensitive Keap1 is covalently modified at specific cysteine residues. Consequently, the latent transcription factor Nrf2 is stabilized and translocates into the nucleus, where it transactivates the expression of detoxification genes through binding to the antioxidant response element (ARE). In the pursuit of potent and bioavailable activators of the ARE, we validated hits from a pathway-directed high-throughput screening campaign by testing them in cell culture and a reporter strain of a whole animal model, Caenorhabditis elegans. These studies allowed us to identify AI-3 as an ARE activator that induces cytoprotective genes in human cells and in worms, which also translated into in vivo activity in mice. AI-3 is an electrophilic ARE activator with two thiol sensitive sites toward a nucleophilic aromatic substitution, and SAR studies indicated the tunability of the system. Tandem LC–MS analysis revealed that AI-3 alkylates Keap1 primarily at Cys151, while AI-3 is reactive toward additional cysteine residues at higher doses in vitro and in vivo. The immediate effects of such alkylation included the disruption of Keap1-Cul3 (low [AI-3]) and/or Keap1-Nrf2 (high [AI-3]) interactions that both led to the stabilization of Nrf2. This further translated into the downstream Nrf2-ARE regulated cytoprotective gene activation. Collectively, AI-3 may become a valuable biological tool and may even provide therapeutic benefits in oxidative stress related diseases.
Co-reporter:Jiyong Hong and Hendrik Luesch
Natural Product Reports 2012 vol. 29(Issue 4) pp:449-456
Publication Date(Web):14 Feb 2012
DOI:10.1039/C2NP00066K
Covering up to 2011
The cyclic depsipeptide largazole from a cyanobacterium of the genus Symploca is a marine natural product with a novel chemical scaffold and potently inhibits class I histone deacetylases (HDACs). Largazole possesses highly differential growth-inhibitory activity, preferentially targeting transformed over non-transformed cells. The intriguing structure and biological activity of largazole have attracted strong interest from the synthetic chemistry community to establish synthetic routes to largazole and to investigate its potential as a cancer therapeutic. This Highlight surveys recent advances in this area with a focus on the discovery, synthesis, target identification, structure–activity relationships, HDAC8–largazole thiol crystal structure, and biological studies, including in vivo anticancer and osteogenic activities.
Co-reporter:Yanxia Liu ; Wei Zhang ; Li Li ; Lilibeth A. Salvador ; Tiantian Chen ; Wuyan Chen ; Kevin M. Felsenstein ; Thomas B. Ladd ; Ashleigh R. Price ; Todd E. Golde ; Jianhua He ; Yechun Xu ; Yingxia Li
Journal of Medicinal Chemistry 2012 Volume 55(Issue 23) pp:10749-10765
Publication Date(Web):November 26, 2012
DOI:10.1021/jm301630s
Inspired by marine cyanobacterial natural products, we synthesized modified peptides with a central statine-core unit, characteristic for aspartic protease inhibition. A series of tasiamide B analogues inhibited BACE1, a therapeutic target in Alzheimer’s disease. We probed the stereospecificity of target engagement and determined additional structure–activity relationships with respect to BACE1 and related aspartic proteases, cathepsins D and E. We cocrystallized selected inhibitors with BACE1 to reveal the structural basis for the activity. Hybrid molecules that combine features of tasiamide B and an isophthalic acid moiety-containing sulfonamide showed nanomolar cellular activity. Compounds were screened in a series of rigorous complementary cell-based assays. We measured secreted APP ectodomain (sAPPβ), membrane bound carboxyl terminal fragment (CTF), levels of β-amyloid (Aβ) peptides and selectivity for β-secretase (BACE1) over γ-secretase. Prioritized compounds showed reasonable stability in vitro and in vivo, and our most potent inhibitor showed efficacy in reducing Aβ levels in the rodent brain.
Co-reporter:Siming Yang, Wei Zhang, Ning Ding, Jeannette Lo, Yanxia Liu, Michael J. Clare-Salzler, Hendrik Luesch, Yingxia Li
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 15) pp:4774-4780
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmc.2012.05.077
The linear depsipeptide grassystatin A, a valuable probe for the study of cathepsin E function, has been synthesized by a [4+6] strategy. It exhibited specific inhibitory activity against cathepsin E with an IC50 value of 0.8 nM. Our studies indicated that inhibition of cathepsin E did not have an impact on ovalbumin antigen processing and peptide presentation, unique from studies of other aspartic protease inhibitors.The linear depsipeptide grassystatin A, a valuable probe for the study of cathepsin E function, has been synthesized by a [4+6] strategy. It exhibited specific inhibitory activity against cathepsin E with an IC50 value of 0.8 nM. And our studies indicated that inhibition of cathepsin E did not have an impact on ovalbumin antigen processing and peptide presentation, unique from studies of other aspartic protease inhibitors.
Co-reporter:Rana Montaser;Dr. Valerie J. Paul; Hendrik Luesch
ChemBioChem 2012 Volume 13( Issue 18) pp:2676-2681
Publication Date(Web):
DOI:10.1002/cbic.201200502
Co-reporter:Lilibeth A. Salvador, Jason S. Biggs, Valerie J. Paul, and Hendrik Luesch
Journal of Natural Products 2011 Volume 74(Issue 5) pp:917-927
Publication Date(Web):March 29, 2011
DOI:10.1021/np200076t
Cytotoxicity-directed purification of a Symploca cf. hydnoides sample from Cetti Bay, Guam, afforded seven new cyclic depsipeptides, veraguamides A−G (1−7), together with the known compound dolastatin 16. The planar structures of 1−7 were elucidated using NMR and MS experiments, while enantioselective HPLC and Mosher’s analysis of acid and base hydrolysates, respectively, were utilized to assign the absolute configurations of the stereocenters. Veraguamides A−G (1−7) are characterized by the presence of an invariant proline residue, multiple N-methylated amino acids, an α-hydroxy acid, and a C8-polyketide-derived β-hydroxy acid moiety with a characteristic terminus as either an alkynyl bromide, alkyne, or vinyl group. These compounds and a semisynthetic analogue (8) showed moderate to weak cytotoxic activity against HT29 colorectal adenocarcinoma and HeLa cervical carcinoma cell lines. Preliminary structure−activity relationship analysis identified several sensitive positions in the veraguamide scaffold that affect the cytotoxic activity of this compound class. Dolastatin 16 showed only weak cytotoxic activity on both cell lines tested. The complete stereostructure of dolastatin 16 was proposed for the first time through degradation followed by a combination of advanced Marfey’s analysis and modified Mosher’s analysis using phenylglycine methyl ester as a chiral anisotropic reagent.
Co-reporter:Rana Montaser, Khalil A. Abboud, Valerie J. Paul, and Hendrik Luesch
Journal of Natural Products 2011 Volume 74(Issue 1) pp:109-112
Publication Date(Web):December 7, 2010
DOI:10.1021/np1006839
An unusual cyclic depsipeptide, pitiprolamide (1), was isolated from the marine cyanobacterium Lyngbya majuscula collected at Piti Bomb Holes, Guam. The structure was deduced using NMR, MS, X-ray crystallography, and enantioselective HPLC-MS techniques. Remarkably, proline represents half of the residues forming pitiprolamide (1). Other distinctive features include a 4-phenylvaline (dolaphenvaline, Dpv) moiety initially found in dolastatin 16 and the rare 2,2-dimethyl-3-hydroxyhexanoic acid (Dmhha) unit condensed in a unique sequence in one single molecule. Pitiprolamide (1) showed weak cytotoxic activity against HCT116 colon and MCF7 breast cancer cell lines, as well as weak antibacterial activities against Mycobacterium tuberculosis and Bacillus cereus.
Co-reporter:Qi-Yin Chen, Yanxia Liu, and Hendrik Luesch
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 11) pp:861
Publication Date(Web):August 31, 2011
DOI:10.1021/ml200176m
Apratoxins are cytotoxic marine natural products that prevent cotranslational translocation early in the secretory pathway. We showed that apratoxins downregulate receptors and growth factor ligands, giving a one–two punch to cancer cells, particularly those that rely on autocrine loops. Through total synthesis, we tested the effects of amino acid substitutions, including alanine scanning, on the downregulation of receptor tyrosine kinases and vascular endothelial growth factor A (VEGF-A) and probed the stereospecificity of target engagement by epimerization of selected chiral centers. Differential effects on two types of secretory molecules suggest that the apratoxins' substrate selectivity with respect to inhibition of secretion may be tuned through structural modifications to provide tailored therapy. Our structure–activity relationship studies and medicinal chemistry efforts led to a potent inhibitor with in vivo efficacy in a colorectal tumor xenograft model without irreversible toxicity exerted by apratoxin A, demonstrating that this novel mechanism of action has therapeutic potential.Keywords: Antitumor agents; growth factors; natural products; receptor tyrosine kinases; secretory pathway; total synthesis
Co-reporter:Jason Christopher Kwan, Theresa Meickle, Dheran Ladwa, Max Teplitski, Valerie Paul and Hendrik Luesch
Molecular BioSystems 2011 vol. 7(Issue 4) pp:1205-1216
Publication Date(Web):24 Jan 2011
DOI:10.1039/C0MB00180E
Quorum sensing (QS) is a mechanism of bacterial gene regulation in response to increases in population density. Perhaps most studied are QS pathways mediated by acylhomoserine lactones (AHLs) in Gram-negative bacteria. Production of small molecule QS signals, their accumulation within a diffusion-limited environment and their binding to a LuxR-type receptor trigger QS-controlled gene regulatory cascades. In Pseudomonas aeruginosa, for example, binding of AHLs to their cognate receptors (LasR, RhlR) controls production of virulence factors, pigments, antibiotics and other behaviors important for its interactions with eukaryotic hosts and other bacteria. We have previously shown that marine cyanobacteria produce QS-inhibitory molecules, including 8-epi-malyngamide C (1), malyngamide C (2) and malyngolide (3). Here we isolated a new small cyclopropane-containing fatty acid, lyngbyoic acid (4), as a major metabolite of the marine cyanobacterium, Lyngbya cf. majuscula, collected at various sites in Florida. We screened 4 against four reporters based on different AHL receptors (LuxR, AhyR, TraR and LasR) and found that 4 most strongly affected LasR. We also show that 4 reduces pyocyanin and elastase (LasB) both on the protein and transcript level in wild-type P. aeruginosa, and that 4 directly inhibits LasB enzymatic activity. Conversely, dodecanoic acid (9) increased pyocyanin and LasB, demonstrating that the fused cyclopropane “tag” is functionally relevant and potentially confers resistance to β-oxidation. Global transcriptional effects of 4 in some ways replicate the gene expression changes of P. aeruginosa during chronic lung infections of cystic fibrosis patients, with reduced lasR signaling, increased biofilm and expression of the virulence locus HSI-I. Compound 4 may therefore prove to be a useful tool in the study of P. aeruginosa adaption during such chronic infections.
Co-reporter:Rana Montaser, Valerie J. Paul, Hendrik Luesch
Phytochemistry 2011 Volume 72(Issue 16) pp:2068-2074
Publication Date(Web):November 2011
DOI:10.1016/j.phytochem.2011.07.014
Pitipeptolides A (1) and B (2) are cyclic depsipeptides isolated from the marine cyanobacterium Lyngbya majuscula from Piti Bomb Holes, Guam. Additional analogues have now been isolated by revisiting larger collections of the same cyanobacterium. The four identified analogues, pitipeptolides C–F (3–6), are the tetrahydro analogue (3), an analogue with a lower degree of methylation (4) as well as two homologues (5 and 6) of pitipeptolide A. Their structures were elucidated using 2D NMR experiments, chiral HPLC analysis and comparison with pitipeptolide A. The identified analogues showed weaker cytotoxic activities compared to the two major parent compounds, pitipeptolides A (1) and B (2), against HT-29 colon adenocarcinoma and MCF7 breast cancer cells. On the other hand, pitipeptolide F (6) was the most potent pitipeptolide in a disc diffusion assay against Mycobacterium tuberculosis. The latter finding suggests that the structure of pitipeptolides could be optimized for selective antibacterial activity.Graphical abstractFurther chemical investigation of a marine cyanobacterium that produces pitipeptolides A and B led to identification of pitipeptolides C–F. These analogues showed weaker cytotoxic activities against cancer cells compared to the two originally identified major compounds. However, pitipeptolide F was the most potent antimycobacterial compound among this series, thus demonstrating selective biological activities among the pitipeptolides.Highlights► Pitipeptolides C–F are identified from a marine cyanobacterium from Guam. ► Pitipeptolides possess weak cytotoxic activity against cancer cell lines. ► Pitipeptolides possess moderate antimycobacterial activity. ► Structural requirements for anticancer and antibacterial activities are different.
Co-reporter:Justin B. Wenger;Napoleon Santos;Yanxia Liu;Jennifer Dallas
Oncology Reviews 2011 Volume 5( Issue 3) pp:
Publication Date(Web):2011 September
DOI:10.1007/s12156-011-0082-3
Antiangiogenic therapy has shown promise in the treatment of patients with hepatocellular carcinoma (HCC). Bevacizumab, sorafenib, and sunitinib showed efficacy in patients with HCC; and sorafenib is approved by the FDA for treatment of this cancer. In practice, the clinical benefit of these agents has been heterogeneous; and in patients who do respond, the benefit is modest and/or short-lived. Recent advances in the molecular understanding of tumor angiogenesis along with the rapid development of targeted drug discovery have made it possible to explore novel combination therapy for HCC. We review the clinical trial results, discuss possible molecular mechanisms of resistance, and suggest novel combinations with antiangiogenic therapy.
Co-reporter:Wooyoung Hur, Zheng Sun, Tao Jiang, Daniel E. Mason, Eric C. Peters, Donna D. Zhang, Hendrik Luesch, Peter G. Schultz, Nathanael S. Gray
Chemistry & Biology 2010 Volume 17(Issue 5) pp:537-547
Publication Date(Web):28 May 2010
DOI:10.1016/j.chembiol.2010.03.013
Eukaryotic cells counteract oxidative and other environmental stress through the activation of Nrf2, the transcription factor that controls the expression of a host of protective enzymes by binding to the antioxidant response element (ARE). The electrophilic molecules that are able to activate Nrf2 and its downstream target genes have demonstrated therapeutic potential in carcinogen-induced tumor models. Using a high-throughput cellular screen, we discovered a class of ARE activator, which we named AI-1, that activates Nrf2 by covalently modifying Keap1, the negative regulator of Nrf2. Biochemical studies indicated that modification of Cys151 of Keap1 by AI-1 disrupted the ability of Keap1 to serve as an adaptor for Cul3-Keap1 ubiquitin ligase complex, thereby causing stabilization and transcriptional activation of Nrf2. AI-1 and its biotinylated derivative are useful pharmacological probes for investigating the molecular details of the cellular antioxidant response.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (231 K)Download as PowerPoint slideHighlights► Keap1-Nrf2 pathway is involved in cellular protection against oxidative stress ► Electrophilic small molecules that stimulate this pathway possess therapeutic benefit under stress-related conditions and diseases ► A new class of Nrf2 activating electrophile (AI-1) was identified from small-molecule screening ► AI-1 undergoes a nucleophilic aromatic substitution reaction with multiple cysteine residues of the cellular redox sensor Keap1 ► Modification of Keap1 Cys151 by AI-1 disrupts the ability of Keap1 to serve as an adaptor for Cul3-ubiquitin E3 ligase complex, thereby leading to Nrf2 activation
Co-reporter:Susan Matthew, Lilibeth A. Salvador, Peter J. Schupp, Valerie J. Paul, and Hendrik Luesch
Journal of Natural Products 2010 Volume 73(Issue 9) pp:1544-1552
Publication Date(Web):August 12, 2010
DOI:10.1021/np1004032
Collections of the marine cyanobacterium Lyngbya bouillonii from shallow patch reefs in Apra Harbor, Guam, afforded three hitherto undescribed analogues of the glycosidic macrolide lyngbyaloside, namely, 2-epi-lyngbyaloside (1) and the regioisomeric 18E- and 18Z-lyngbyalosides C (2 and 3). Concurrently we discovered two new analogues of the cytoskeletal actin-disrupting lyngbyabellins, 27-deoxylyngbyabellin A (4) and lyngbyabellin J (5), a novel macrolide of the laingolide family, laingolide B (6), and a linear modified peptide, lyngbyapeptin D (7), along with known lyngbyabellins A and B, lyngbyapeptin A, and lyngbyaloside. The structures of 1−7 were elucidated by a combination of NMR spectroscopic and mass spectrometric analysis. Compounds 1−6 were either brominated (1−3) or chlorinated (4−6), consistent with halogenation being a hallmark of many marine natural products. All extracts derived from these L. bouillonii collections were highly cytotoxic due to the presence of apratoxin A or apratoxin C. Compounds 1−5 showed weak to moderate cytotoxicity to HT29 colorectal adenocarcinoma and HeLa cervical carcinoma cells.
Co-reporter:Jason C. Kwan, Max Teplitski, Sarath P. Gunasekera, Valerie J. Paul and Hendrik Luesch
Journal of Natural Products 2010 Volume 73(Issue 3) pp:463-466
Publication Date(Web):February 18, 2010
DOI:10.1021/np900614n
A new stereoisomer of malyngamide C, 8-epi-malyngamide C (1), and the known compound lyngbic acid [(4E,7S)-7-methoxytetradec-4-enoic acid] were isolated from a sample of Lyngbya majuscula collected near Bush Key, Dry Tortugas, Florida. The structure of 1 was determined by NMR and MS experiments. The absolute configuration of 1 was determined by selective Mitsunobu inversion of C-8 to give malyngamide C, as determined by NMR, MS, and comparison of specific rotation. Both 1 and malyngamide C were found to be cytotoxic to HT29 colon cancer cells (IC50 15.4 and 5.2 μM, respectively) and to inhibit bacterial quorum sensing in a reporter gene assay.
Co-reporter:Lilibeth A. Salvador, Valerie J. Paul, and Hendrik Luesch
Journal of Natural Products 2010 Volume 73(Issue 9) pp:1606-1609
Publication Date(Web):August 31, 2010
DOI:10.1021/np100467d
A Phormidium spp. collection from Key West, Florida, afforded caylobolide B (1), an analogue of the known macrolactone caylobolide A, previously isolated from a Lyngbya majuscula collection from the Bahamas. The planar structure of 1 was determined using NMR and MS experiments. The relative configuration for subunits C7−C9 and C25−C29 was assigned using Kishi’s Universal NMR Database. Caylobolide B (1) displayed cytotoxic activity against HT29 colorectal adenocarcinoma and HeLa cervical carcinoma cells with IC50 values of 4.5 and 12.2 μM, respectively.
Co-reporter:Jason C. Kwan, Ranjala Ratnayake, Khalil A. Abboud, Valerie J. Paul, and Hendrik Luesch
The Journal of Organic Chemistry 2010 Volume 75(Issue 23) pp:8012-8023
Publication Date(Web):November 4, 2010
DOI:10.1021/jo1013564
Grassypeptolides A−C (1−3), a group of closely related bis-thiazoline containing cyclic depsipeptides, have been isolated from extracts of the marine cyanobacterium Lyngbya confervoides. Although structural differences between the analogues are minimal, comparison of the in vitro cytotoxicity of the series revealed a structure−activity relationship. When the ethyl substituent of 1 is changed to a methyl substituent in 2, activity is only slightly reduced (3−4-fold), whereas inversion of the Phe unit flanking the bis-thiazoline moiety results in 16−23-fold greater potency. We show that both 1 and 3 cause G1 phase cell cycle arrest at lower concentrations, followed at higher concentrations by G2/M phase arrest, and that these compounds bind Cu2+ and Zn2+. The three-dimensional structure of 2 was determined by MS, NMR, and X-ray crystallography, and the structure of 3 was established by MS, NMR, and chemical degradation. The structure of 3 was explored by in silico molecular modeling, revealing subtle differences in overall conformation between 1 and 3. Attempts to interconvert 1 and 3 with base were unsuccessful, but enzymatic conversion may be possible and could be a novel form of activation for chemical defense.
Co-reporter:Jason C. Kwan ;Dr. Hendrik Luesch
Chemistry - A European Journal 2010 Volume 16( Issue 44) pp:13020-13029
Publication Date(Web):
DOI:10.1002/chem.201001562
Abstract
Bioactive natural products often possess uniquely functionalized structures with unusual modes of action; however, the natural product itself is not always the active species. We discuss molecules that draw on protecting group chemistry or else require activation to unmask reactive centers, illustrating that nature is not only a source of complex structures but also a guide for elegant chemical transformations which provides ingenious chemical solutions for drug delivery.
Co-reporter:Jason C. Kwan ; Erika A. Eksioglu ; Chen Liu ; Valerie J. Paul
Journal of Medicinal Chemistry 2009 Volume 52(Issue 18) pp:5732-5747
Publication Date(Web):August 28, 2009
DOI:10.1021/jm9009394
In our efforts to explore marine cyanobacteria as a source of novel bioactive compounds, we discovered a statine unit-containing linear decadepsipeptide, grassystatin A (1), which we screened against a diverse set of 59 proteases. We describe the structure determination of 1 and two natural analogues, grassystatins B (2) and C (3), using NMR, MS, and chiral HPLC techniques. Compound 1 selectively inhibited cathepsins D and E with IC50s of 26.5 nM and 886 pM, respectively. Compound 2 showed similar potency and selectivity against cathepsins D and E (IC50s of 7.27 nM and 354 pM, respectively), whereas the truncated peptide analogue grassystatin C (3), which consists of two fewer residues than 1 and 2, was less potent against both but still selective for cathepsin E. The selectivity of compounds 1−3 for cathepsin E over D (20−38-fold) suggests that these natural products may be useful tools to probe cathepsin E function. We investigated the structural basis of this selectivity using molecular docking. We also show that 1 can reduce antigen presentation by dendritic cells, a process thought to rely on cathepsin E.
Co-reporter:Susan Matthew, Valerie J. Paul, Hendrik Luesch
Phytochemistry 2009 70(17–18) pp: 2058-2063
Publication Date(Web):
DOI:10.1016/j.phytochem.2009.09.010
Co-reporter:Kanchan Taori;Yanxia Liu Dr.;Valerie J. Paul Dr. Dr.
ChemBioChem 2009 Volume 10( Issue 10) pp:1634-1639
Publication Date(Web):
DOI:10.1002/cbic.200900192
Co-reporter:Susan Matthew, Peter J. Schupp and Hendrik Luesch
Journal of Natural Products 2008 Volume 71(Issue 6) pp:1113-1116
Publication Date(Web):May 8, 2008
DOI:10.1021/np700717s
A collection of the marine cyanobacterium Lyngbya bouillonii from Guam afforded apratoxin E (1), a new peptide−polyketide hybrid of the apratoxin class of cytotoxins. The planar structure of 1 was elucidated by NMR spectroscopic analysis and mass spectrometry. Configurational assignments of stereocenters in the peptide portion were made by chiral HPLC analysis of the acid hydrolysate. The relative configuration in the polyketide moiety was assigned by comparison of NMR data including proton−proton coupling constants with those of the known analogues. Apratoxin E (1) displayed strong cytotoxicity against several cancer cell lines derived from colon, cervix, and bone, ranging from 21 to 72 nM, suggesting that the α,β-unsaturation of the modified cysteine residue is not essential for apratoxin activity. The 5- to 15-fold reduced activity compared with apratoxin A (2) is attributed to the dehydration in the long-chain polyketide unit, which could affect the conformation of the molecule.
Co-reporter:Kanchan Taori, Valerie J. Paul and Hendrik Luesch
Journal of Natural Products 2008 Volume 71(Issue 9) pp:1625-1629
Publication Date(Web):August 12, 2008
DOI:10.1021/np8002172
Two cyclodepsipeptides named kempopeptins A (1) and B (2) were isolated from a collection of a Floridian marine cyanobacterium, Lyngbya sp., that had previously afforded the structurally related potent elastase inhibitors lyngbyastatin 7 and somamide B. The structures of 1 and 2 were elucidated mainly by 1D and 2D NMR spectroscopy, and the absolute configuration was established by chiral HPLC and Marfey’s analysis of the degradation products. Kempopeptin A (1) exhibited an IC50 against elastase of 0.32 μM and against chymotrypsin of 2.6 μM, while kempopeptin B (2) inhibited trypsin with an IC50 of 8.4 μM.
Co-reporter:Kanchan Taori, Susan Matthew, James R. Rocca, Valerie J. Paul and Hendrik Luesch
Journal of Natural Products 2007 Volume 70(Issue 10) pp:1593-1600
Publication Date(Web):October 3, 2007
DOI:10.1021/np0702436
Three new analogues of dolastatin 13, termed lyngbyastatins 5–7 (1–3), were isolated from two different collections of marine cyanobacteria, Lyngbya spp., from South Florida. Their planar structures were deduced by a combination of NMR techniques, and the absolute configurations were established by modified Marfey’s analysis of the acid hydrolyzates. The related cyclodepsipeptide somamide B (4), previously reported from a Fijian cyanobacterium, has also been found in one of the extracts, and its absolute stereochemistry was unambiguously assigned for the first time. Compounds 1–4 were found to selectively inhibit elastase over several other serine proteases, with IC50 values for porcine pancreatic elastase ranging from 3 to 10 nM.
Co-reporter:Yanxia Liu;Jonathan T. Kern;John R. Walker;Jeffrey A. Johnson;Peter G. Schultz
PNAS 2007 104 (12 ) pp:5205-5210
Publication Date(Web):2007-03-20
DOI:10.1073/pnas.0700898104
The antioxidant response element (ARE) is a cis-acting regulatory enhancer element found in the 5′ flanking region of many
phase II detoxification enzymes. Up-regulation of ARE-dependent target genes is known to have neuroprotective effects; yet,
the mechanism of activation is largely unknown. By screening an arrayed collection of ≈15,000 full-length expression cDNAs
in the human neuroblastoma cell line IMR-32 with an ARE-luciferase reporter, we have identified several cDNAs not previously
associated with ARE activation. A subset of cDNAs, encoding sequestosome 1 (SQSTM1) and dipeptidylpeptidase 3 (DPP3), activated
the ARE in primary mouse-derived cortical neurons. Overexpression of SQSTM1 and DPP3 in IMR-32 cells stimulated NF-E2-related
factor 2 (NRF2) nuclear translocation and led to increased levels of NAD(P)H:quinone oxidoreductase 1, a protein which is
transcriptionally regulated by the ARE. When transfected into IMR-32 neuroblastoma cells that were depleted of transcription
factor NRF2 by RNA interference, SQSTM1 and DPP3 were unable to activate the ARE or induce NAD(P)H:quinone oxidoreductase
1 expression, indicating that the ARE activation upon ectopic expression of these cDNAs is mediated by NRF2. Studies with
pharmacological inhibitors indicated that 1-phosphatidylinositol 3-kinase and protein kinase C signaling are essential for
activity. Overexpression of these cDNAs conferred partial resistance to hydrogen peroxide or rotenone-induced toxicity, consistent
with the induction of antioxidant and phase II detoxification enzymes, which can protect from oxidative stress. This work
and other such studies may provide mechanisms for activating the ARE in the absence of general oxidative stress and a yet-unexploited
therapeutic approach to degenerative diseases and aging.
Co-reporter:Hendrik Luesch
Molecular BioSystems 2006 vol. 2(Issue 12) pp:609-620
Publication Date(Web):27 Oct 2006
DOI:10.1039/B609384A
Drug discovery is hampered by the lack of general strategies to characterize the mechanisms of action and intracellular targets of bioactive small molecules. Genomics and proteomics promise to aid in this process. Genome-wide approaches in yeast have proven useful to infer the targets and target pathways of small molecules. These approaches are being systematically transferred into mammalian cell culture systems in order to interrogate more complex pathways in a more relevant setting. Advances in proteomics and in vivo genetic screening in multicellular model organism systems are also becoming increasingly powerful and amenable to high-throughput. Current methodologies and technologies are discussed, including how these global approaches complement affinity-based target identification strategies.

![(3AS,7AS)-1,3-BIS[(4-BROMOPHENYL)METHYL]-2-[(E)-BUT-2-ENYL]-2-CHLORO-3A,4,5,6,7,7A-HEXAHYDROBENZO[D][1,3,2]DIAZASILOLE](http://img.cochemist.com/ccimg/1072300/1072220-37-1.png)
![(3AS,7AS)-1,3-BIS[(4-BROMOPHENYL)METHYL]-2-[(E)-BUT-2-ENYL]-2-CHLORO-3A,4,5,6,7,7A-HEXAHYDROBENZO[D][1,3,2]DIAZASILOLE](http://img.cochemist.com/ccimg/1072300/1072220-37-1_b.png)




















![Heptanal, 5-[(4-methoxyphenyl)methoxy]-3,6,6-trimethyl-, (3R,5S)-](http://img.cochemist.com/ccimg/676400/676324-77-9.png)
![Heptanal, 5-[(4-methoxyphenyl)methoxy]-3,6,6-trimethyl-, (3R,5S)-](http://img.cochemist.com/ccimg/676400/676324-77-9_b.png)
![2-HEXANONE, 4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-5,5-DIMETHYL-, (4S)-](http://img.cochemist.com/ccimg/612500/612492-55-4.png)
![2-HEXANONE, 4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]-5,5-DIMETHYL-, (4S)-](http://img.cochemist.com/ccimg/612500/612492-55-4_b.png)



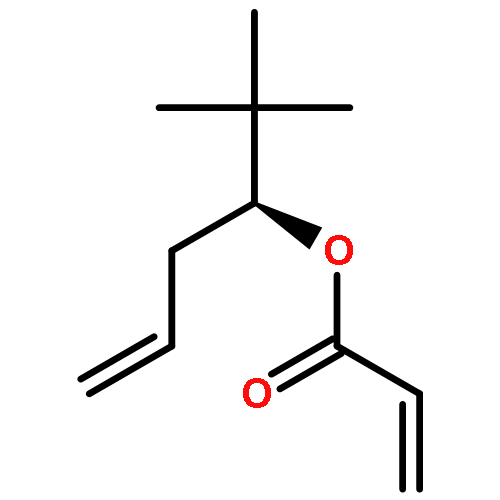
![L-Threonine, N-[(1,1-dimethylethoxy)carbonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/545500/545432-97-1.png)
![L-Threonine, N-[(1,1-dimethylethoxy)carbonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/545500/545432-97-1_b.png)


![L-Norvaline, 5-hydroxy-N-[(2-propenyloxy)carbonyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/500200/500145-43-7.png)
![L-Norvaline, 5-hydroxy-N-[(2-propenyloxy)carbonyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/500200/500145-43-7_b.png)


![1,3,2-Oxazaborolidin-5-one,4-(1-methylethyl)-3-[(4-methylphenyl)sulfonyl]-, (4R)-](http://img.cochemist.com/ccimg/177300/177274-09-8.png)
![1,3,2-Oxazaborolidin-5-one,4-(1-methylethyl)-3-[(4-methylphenyl)sulfonyl]-, (4R)-](http://img.cochemist.com/ccimg/177300/177274-09-8_b.png)
![L-Glutamic acid, N-[(2-propenyloxy)carbonyl]-, 1-(phenylmethyl) ester](http://img.cochemist.com/ccimg/163800/163711-74-8.png)
![L-Glutamic acid, N-[(2-propenyloxy)carbonyl]-, 1-(phenylmethyl) ester](http://img.cochemist.com/ccimg/163800/163711-74-8_b.png)
![tert-butyl N-[(1S)-2-[methoxy(methyl)amino]-2-oxo-1-(tritylsulfanylmethyl)ethyl]carbamate](http://img.cochemist.com/ccimg/158900/158861-38-2.png)
![tert-butyl N-[(1S)-2-[methoxy(methyl)amino]-2-oxo-1-(tritylsulfanylmethyl)ethyl]carbamate](http://img.cochemist.com/ccimg/158900/158861-38-2_b.png)




![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7.png)
![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7_b.png)











![Methyl (5-butyl-1H-benzo[d]imidazol-2-yl)carbamate](http://img.cochemist.com/ccimg/14300/14255-87-9.png)
![Methyl (5-butyl-1H-benzo[d]imidazol-2-yl)carbamate](http://img.cochemist.com/ccimg/14300/14255-87-9_b.png)




![Card-20(22)-enolide,3-[(6-deoxy-3-O-methyl-a-L-glucopyranosyl)oxy]-14-hydroxy-19-oxo-, (3b,5b)-](http://img.cochemist.com/ccimg/1200/1182-87-2.png)
![Card-20(22)-enolide,3-[(6-deoxy-3-O-methyl-a-L-glucopyranosyl)oxy]-14-hydroxy-19-oxo-, (3b,5b)-](http://img.cochemist.com/ccimg/1200/1182-87-2_b.png)


![Bufa-4,20,22-trienolide,3-[(6-deoxy-a-L-mannopyranosyl)oxy]-14-hydroxy-,(3b)-](http://img.cochemist.com/ccimg/500/466-06-8.png)
![Bufa-4,20,22-trienolide,3-[(6-deoxy-a-L-mannopyranosyl)oxy]-14-hydroxy-,(3b)-](http://img.cochemist.com/ccimg/500/466-06-8_b.png)
![(2S,3S)-2-dimethylamino-N-[(2S)-2-[[(2R)-2-methoxy-4-[(2S)-2-[(2R)-1-methoxy-2-methyl-3-oxo-3-[[(1R)-2-phenyl-1-thiazol-2-yl-ethyl]amino]propyl]pyrrolidin-1-yl]-4-oxo-1-sec-butyl-butyl]-methyl-amino]-3-methyl-butanoyl]-3-methyl-pentanamide](http://img.cochemist.com/ccimg/212100/212007-18-6.png)
![(2S,3S)-2-dimethylamino-N-[(2S)-2-[[(2R)-2-methoxy-4-[(2S)-2-[(2R)-1-methoxy-2-methyl-3-oxo-3-[[(1R)-2-phenyl-1-thiazol-2-yl-ethyl]amino]propyl]pyrrolidin-1-yl]-4-oxo-1-sec-butyl-butyl]-methyl-amino]-3-methyl-butanoyl]-3-methyl-pentanamide](http://img.cochemist.com/ccimg/212100/212007-18-6_b.png)
![L-Valinamide,N,N-dimethyl-L-valyl-N-[(1S,2R)-2-methoxy-4-[(2S)-2-[(1R,2R)-1-methoxy-2-methyl-3-oxo-3-[[(1S)-2-phenyl-1-(2-thiazolyl)ethyl]amino]propyl]-1-pyrrolidinyl]-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-](http://img.cochemist.com/ccimg/110500/110417-88-4.png)
![L-Valinamide,N,N-dimethyl-L-valyl-N-[(1S,2R)-2-methoxy-4-[(2S)-2-[(1R,2R)-1-methoxy-2-methyl-3-oxo-3-[[(1S)-2-phenyl-1-(2-thiazolyl)ethyl]amino]propyl]-1-pyrrolidinyl]-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-](http://img.cochemist.com/ccimg/110500/110417-88-4_b.png)