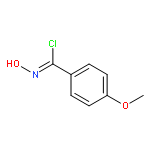
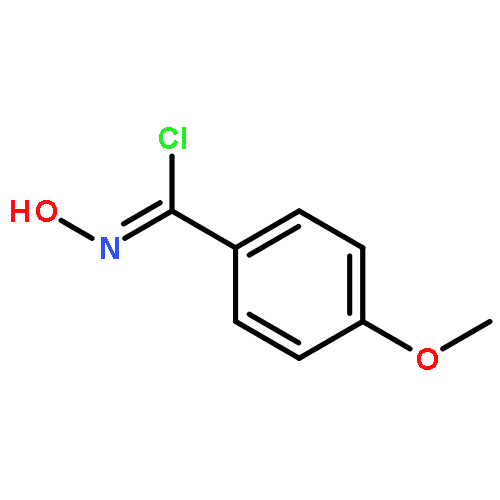
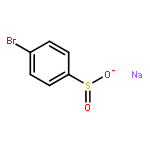
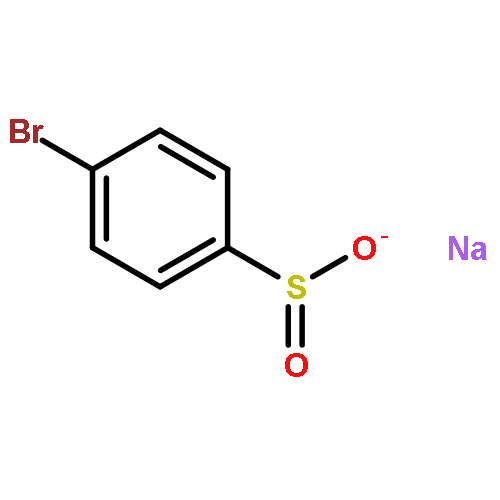
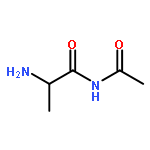
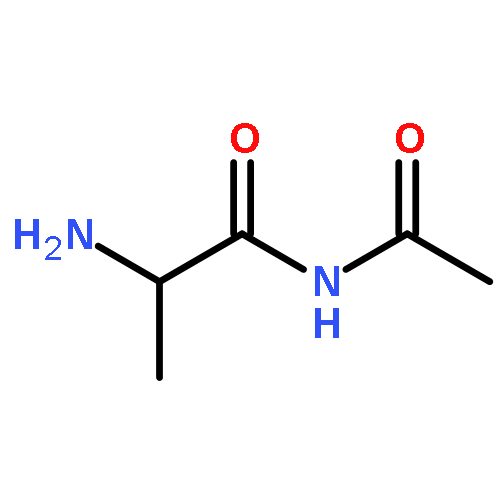
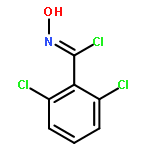
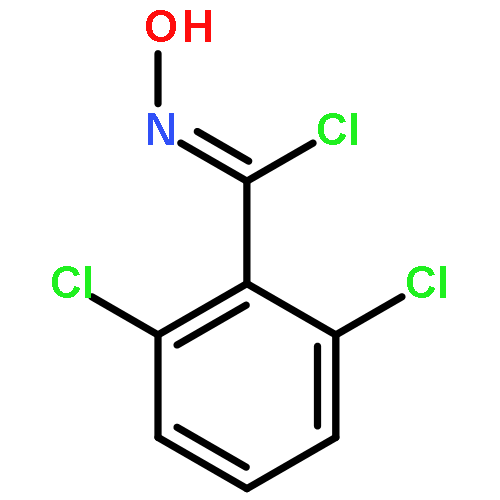
Co-reporter:Yaling Gong, Selin Somersan Karakaya, Xiaoyong Guo, Purong Zheng, Ben Gold, Yao Ma, David Little, Julia Roberts, Thulasi Warrier, Xiuju Jiang, Maneesh Pingle, Carl F. Nathan, Gang Liu
European Journal of Medicinal Chemistry 2014 Volume 75() pp:336-353
Publication Date(Web):21 March 2014
DOI:10.1016/j.ejmech.2014.01.039
•Seventy new benzimidazole-based compounds were designed and synthesized.•All of the compounds were evaluated for activity against replicating and non-replicating Mycobacterium tuberculosis in vitro.•Compounds 49, 67, 68, 69, 70 and 72 exhibited high potency and acceptable selectivity indices.•Several nitrofuranyl benzimidazoles were selectively bactericidal for mycobacteria and killed Mtb in primary human macrophages.•The SOS chromotest indicated that compound 70 had a very low mutagenic potential.Tuberculosis remains one of the deadliest infectious diseases, killing 1.4 million people annually and showing a rapid increase in cases resistant to multiple drugs. New antibiotics against tuberculosis are urgently needed. Here we describe the design, synthesis and structure–activity relationships of a series of benzimidazole-based compounds with activity against Mycobacterium tuberculosis (Mtb) in a replicating state, a physiologically-induced non-replicating state, or both. Compounds 49, 67, 68, 69, 70, and 72, which shared a 5-nitrofuranyl moiety, exhibited high potency and acceptable selectivity indices (SI). As illustrated by compound 70 (MIC90 < 0.049 μg/mL, SI > 512), the 5-nitrofuranyl group was compatible with minimal cytotoxicity and good intra-macrophage killing, although it lacked non-replicating activity when assessed by CFU assays. Compound 70 had low mutagenic potential by SOS Chromotest assay, making this class of compounds good candidates for further evaluation and target identification.A series of novel benzimidazole-based compounds were designed and synthesized. Several nitrofuranyl benzimidazoles are selectively bactericidal for mycobacteria and kill Mtb in primary human macrophages. The most potent compound 70 has a very low mutagenic potential.
Co-reporter:Yao Ma ; Nan Zhao
Journal of Medicinal Chemistry 2011 Volume 54(Issue 8) pp:2767-2777
Publication Date(Web):March 15, 2011
DOI:10.1021/jm101577z
1 (MTC-220), a conjugate of paclitaxel and a muramyl dipeptide analogue, has been synthesized as a novel agent of dual antitumor growth and metastasis activities. In vitro and in vivo tests show that 1 retains its ability to inhibit tumor growth. It is superior to paclitaxel in its ability to prevent tumor metastasis in Lewis lung carcinoma and 4T1-tumor-bearing mice. The present studies indicate that 1 suppresses myeloid derived suppressor cell accumulation in the spleen and bone marrow of tumor-bearing mice and also represses inflammatory cytokines in tumor tissue. These results demonstrated that 1 could be a potential therapeutic and preventive agent for cancer growth and metastasis.
Co-reporter:Rui Liu ; Zhuhui Huang ; Michael G. Murray ; Xiaoyong Guo
Journal of Medicinal Chemistry 2011 Volume 54(Issue 16) pp:5747-5768
Publication Date(Web):July 15, 2011
DOI:10.1021/jm200394x
Hepatitis C virus (HCV) infection is a serious problem worldwide, but no effective drugs are currently available. Through screening of our privileged structure library, quinoxalin-2(1H)-one derivative N-(7-(cyclohexyl(methyl)amino)-3-oxo-3,4-dihydroquinoxalin-6-ylcarbamothioyl)benzamide (compound 1) was identified as potent HCV inhibitor in vitro. Subsequently, a structure–activity relationship analysis was carried out that showed N-(7-(cyclohexyl(methyl)amino)-3-oxo-3,4-dihydroquinoxalin-6-ylcarbamothioyl)furan-2-carboxamide (compound 11, EC50 = 1.8 μM, SI = 9.6), 6-(cyclohexyl(methyl)amino)-7-(4-phenylthiazol-2-ylamino)quinoxalin-2(1H)-one (compound 33, EC50 = 1.67 μM, SI = 37.4), 2-(cyclohexyl(methyl)amino)-3-(4-phenylthiazol-2-ylamino)-7,8,9,10-tetrahydro-5H-pyrido[1,2-a]quinoxalin-6(6aH)-one (compound 60, EC50 = 1.19 μM, SI = 9.27), 8-(cyclohexyl(methyl)amino)-7-(4-phenylthiazol-2-ylamino)pyrrolo[1,2-a]quinoxalin-4(5H)-one (compound 65, EC50 = 1.82 μM, SI = 9.9), and 6-(diethylamino)-7-(4-phenylthiazol-2-ylamino)quinoxalin-2(1H)-one (compound 78, EC50 = 1.27 μM, SI = 17.9) acted against HCV. The data from the structure–activity relationship study suggests that quinoxalin-2(1H)-one derivatives exhibited potent activity against HCV.
Co-reporter:Nan Zhao, Yao Ma, Gang Liu
Chinese Chemical Letters 2011 Volume 22(Issue 12) pp:1443-1446
Publication Date(Web):December 2011
DOI:10.1016/j.cclet.2011.07.020
The solution-phase synthesis of a muramyl dipeptide (MDP) analogue of Nα-[4-chlorocinnamoyl-l-alanyl-d-isoglutaminyl]-l-lysine (MDA, 2) is reported that possesses the features of easy feasibility, safety and low cost in large scale of synthesis.
Co-reporter:Yu Yan, Zijie Liu, Jianjun Zhang, Ruiming Xu, Xiao Hu, Gang Liu
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 14) pp:4189-4192
Publication Date(Web):15 July 2011
DOI:10.1016/j.bmcl.2011.05.080
A series of 2-[(2-pyridylmethyl)sulfinyl]benzimidazole derivatives were synthesized via a solution phase synthetic route using a reversal method of diversity introduction. Using this synthetic strategy, we obtained two key intermediates (4-A and 4-B) simultaneously, which allows us to introduce diversity points onto the benzimidazole part of the final product under reliable reaction conditions to identify potent H+/K+-ATP enzyme inhibitors. Compound 14l (IC50 = 1.6 × 10−5 M) was comparable with H+/K+-ATP enzyme inhibitor in vitro.We have devolped a reverse method to synthesize benzimidazole derivatives that inhibits H+/K+-ATP enzymatic activity as potential proton pump inhibitors.
Co-reporter:Wu Liang
Chinese Journal of Chemistry 2011 Volume 29( Issue 5) pp:983-990
Publication Date(Web):
DOI:10.1002/cjoc.201190200
Abstract
Three efficient methods to synthesize mono- and di-fluorinated benzimidazoles are reported. These methods provide 5-amino-6-fluoro-benzimidazoles (5), 5,7-difluoro-benzimidazoles (10), and 6-fluoro-benzimidazoles (13) starting from commercially available 1,5-difluoro-2,4-dinitrobenzene (DFDNB), 2,3,4,5-tetrafluoro-6-nitrobenzoic acid (TFNBA), and 2,4-difluoro-1-nitrobenzene (DFNB), respectively.
Co-reporter:Chunyan Han;Jinlan Zhang;Mingyue Zheng;Yao Xiao;Yan Li
Molecular Diversity 2011 Volume 15( Issue 4) pp:857-876
Publication Date(Web):2011 November
DOI:10.1007/s11030-011-9317-2
The concept of drug-likeness has been widely applied in combinatorial chemistry as an approach to reduce attrition in drug discovery and development. Meanwhile, bicyclic privileged structures with versatile binding properties have emerged as ideal source of core scaffolds for the design and synthesis of combinatorial libraries. For the purpose of better assisting the design of bicyclic privileged structure-based combinatorial libraries, we conducted an integrated drug-likeness study on compounds of these scaffolds. Distributions of physicochemical properties (PCPs) were analyzed and in silico prediction models were built. Our results showed that there exist much difference between the drug-like ranges (DLRs) of bicyclic privileged structures and that of others, which have significant impact on compound selection. The DLRs for bicyclic privileged structures were defined as 260 ≤ MW ≤ 524; 0.9 ≤ ALogP ≤ 5.4; 2 ≤ Hacc ≤ 8; Hdon ≤ 3; 21.0 ≤ PSA ≤ 128.6; 6.3 ≤ FPSA ≤ 34.2; 1 ≤ RotB ≤ 10; 2 ≤ Nr ≤ 5; 1 ≤ Nc ≤ 7; SA ≤ 4. Two accurate and easy to understand in silico prediction models, Caco-2 permeability model and metabolic stability classification model, had been built to guide drug candidate optimization. In these models, hydrogen-bond donor and rotatable bond showed major impact on the permeability of compounds, while lipophilicity, flexibility, degree of branching and the existence of some functional groups determined the fate of a drug in metabolic process. Suggestions on structural modification toward higher permeability and metabolic stability were given according to the in silico models.
Co-reporter:Hai Xue ; Xiaofan Lu ; Purong Zheng ; Li Liu ; Chunyan Han ; Jinping Hu ; Zijie Liu ; Tao Ma ; Yan Li ; Lin Wang ; Zhiwei Chen
Journal of Medicinal Chemistry 2010 Volume 53(Issue 3) pp:1397-1401
Publication Date(Web):January 5, 2010
DOI:10.1021/jm901653e
We herein report a new compound: 10-chloromethyl-11-demethyl-12-oxo-calanolide A (20, EC50 = 7.4 nM, SI = 1417), which demonstrates a druggable profile with 32.7% oral bioavailability in rat, tolerated oral single dose toxicity in mice, and especially the feature of highly efficient suppression of the wild-type HIV-1 and Y181C mutant HIV-1 at an EC50 = 7.4 nM and EC50 = 0.46 nM, respectively.
Co-reporter:Gang Li ; Dongmei Wang ; Mingna Sun ; Guangyan Li ; Jinfeng Hu ; Yun Zhang ; Yuhe Yuan ; Haijie Ji ; Naihong Chen
Journal of Medicinal Chemistry 2010 Volume 53(Issue 4) pp:1741-1754
Publication Date(Web):January 25, 2010
DOI:10.1021/jm901652p
Chemokine-like factor 1 (CKLF1) is a novel functional cytokine that acts through its receptor CC chemokine receptor 4 (CCR4). Activation of CCR4 by CKLF1 plays an important role in diseases such as asthma and multiple sclerosis. This article describes a cell-based screening assay using an FITC-labeled CCR4 agonist (CKLF1-C27), a CKLF1 peptide fragment. Screening of our in-stock small-molecule library identified a 3-piperazinylcoumarin analogue 1 (IC50 = 4.36 × 10−6 M) that led to the discovery of orally active compound 41 (IC50 = 2.12 × 10−8 M) through systematic optimization. Compound 41 blocked the calcium mobilization and chemotaxis induced by CKLF1-C27 and reduced the asthmatic pathologic changes in lung tissue of human CKLF1-transfected mice. Further studies indicated that compound 41 ameliorated pathological changes via inhibition of the NF-κB signal pathway.
Co-reporter:Hao Wu, Xilei Xie and Gang Liu
ACS Combinatorial Science 2010 Volume 12(Issue 3) pp:346
Publication Date(Web):March 2, 2010
DOI:10.1021/cc900173s
Three diversity points of 4(3H)-quinazolinone are introduced at the 3-, 6-, and 7-positions with an efficient parallel solution-phase synthetic method. A one-pot synthesis was developed that gave the key intermediate in high yield. Five hit compounds exhibit preferable activities against a panel of human tumor cell lines, which pointed out preliminary structure−activity relationships.
Co-reporter:Ning Zhu, Fa Zhang and Gang Liu
ACS Combinatorial Science 2010 Volume 12(Issue 4) pp:531
Publication Date(Web):May 28, 2010
DOI:10.1021/cc100042v
The thiol-disulfide dynamic interchange reaction mediated by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was extensively studied. By this synthetic method sulfides can be prepared successfully within seconds in high yields at room temperature from stable and readily available disulfides and an alkylating agent. The method was further demonstrated to efficiently produce benzofused nitrogen−sulfur heterocycles with high skeletal diversity in a one-pot process.
Co-reporter:Hai Xue, Tao Ma, Lin Wang, Gang Liu
Chinese Chemical Letters 2010 Volume 21(Issue 11) pp:1291-1294
Publication Date(Web):November 2010
DOI:10.1016/j.cclet.2010.04.030
An unexpected reaction of 10-bromomethyl-12-oxocalanolide A (2) with primary amine was demonstrated and studied. Consequentially, a new method for the preparation of N-substituted pyrroles starting from γ-halo-α,β-unsaturated ketone was presented.



























![[1,1'-BIPHENYL]-3-CARBONITRILE, 4-AMINO-3'-NITRO-](http://img.cochemist.com/ccimg/405600/405520-52-7.png)
![[1,1'-BIPHENYL]-3-CARBONITRILE, 4-AMINO-3'-NITRO-](http://img.cochemist.com/ccimg/405600/405520-52-7_b.png)
























![Benzofuran, 2-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/153600/153561-81-0.png)
![Benzofuran, 2-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/153600/153561-81-0_b.png)
![2H,6H,10H-Dipyrano[2,3-f:2',3'-h][1]benzopyran-2-one,11,12-dihydro-12-hydroxy-6,6,10,11-tetramethyl-4-propyl-, (10R,11S,12S)-](http://img.cochemist.com/ccimg/142700/142632-32-4.png)
![2H,6H,10H-Dipyrano[2,3-f:2',3'-h][1]benzopyran-2-one,11,12-dihydro-12-hydroxy-6,6,10,11-tetramethyl-4-propyl-, (10R,11S,12S)-](http://img.cochemist.com/ccimg/142700/142632-32-4_b.png)




![Methyl 4-[(z)-c-chloro-n-hydroxycarbonimidoyl]benzoate](http://img.cochemist.com/ccimg/101100/101023-70-5.png)
![Methyl 4-[(z)-c-chloro-n-hydroxycarbonimidoyl]benzoate](http://img.cochemist.com/ccimg/101100/101023-70-5_b.png)
![3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin-10(3H)-one,octahydro-3,6-dimethyl-9-methylene-, (3R,5aS,6R,8aS,12S,12aR)-](http://img.cochemist.com/ccimg/101100/101020-89-7.png)
![3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin-10(3H)-one,octahydro-3,6-dimethyl-9-methylene-, (3R,5aS,6R,8aS,12S,12aR)-](http://img.cochemist.com/ccimg/101100/101020-89-7_b.png)













![10aH-9,10b-Epoxypyrano[4,3,2-jk][2]benzoxepin-2(3H)-one,octahydro-3,6,9-trimethyl-, (3R,3aS,6R,6aS,9S,10aS,10bR)-](http://img.cochemist.com/ccimg/72900/72826-63-2.png)
![10aH-9,10b-Epoxypyrano[4,3,2-jk][2]benzoxepin-2(3H)-one,octahydro-3,6,9-trimethyl-, (3R,3aS,6R,6aS,9S,10aS,10bR)-](http://img.cochemist.com/ccimg/72900/72826-63-2_b.png)


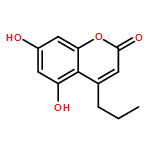

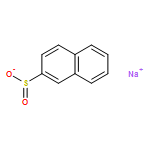
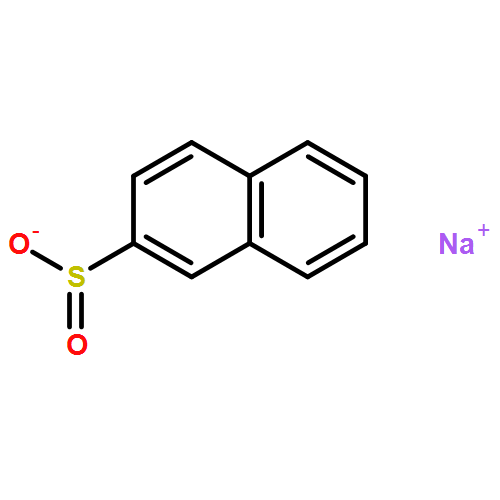
![1-(6-Iodobenzo[d][1,3]dioxol-5-yl)ethanone](http://img.cochemist.com/ccimg/61600/61599-79-9.png)
![1-(6-Iodobenzo[d][1,3]dioxol-5-yl)ethanone](http://img.cochemist.com/ccimg/61600/61599-79-9_b.png)
![[1,1'-Biphenyl]-3-carbonitrile, 4-amino-](http://img.cochemist.com/ccimg/55700/55675-86-0.png)
![[1,1'-Biphenyl]-3-carbonitrile, 4-amino-](http://img.cochemist.com/ccimg/55700/55675-86-0_b.png)