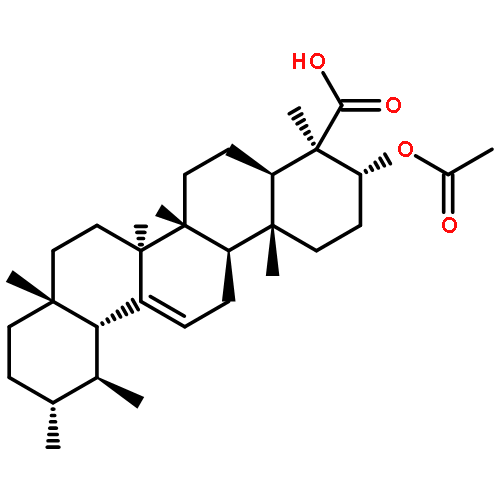
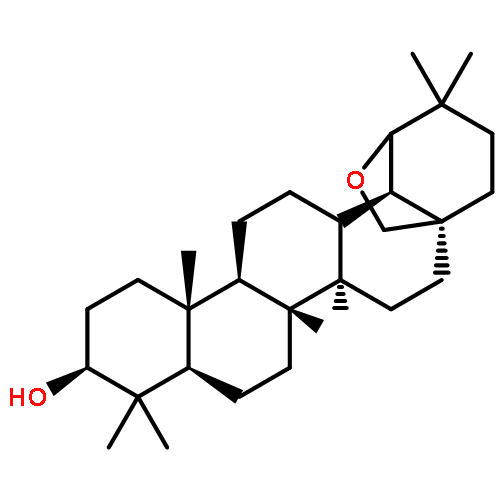
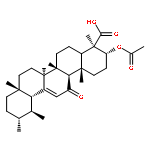
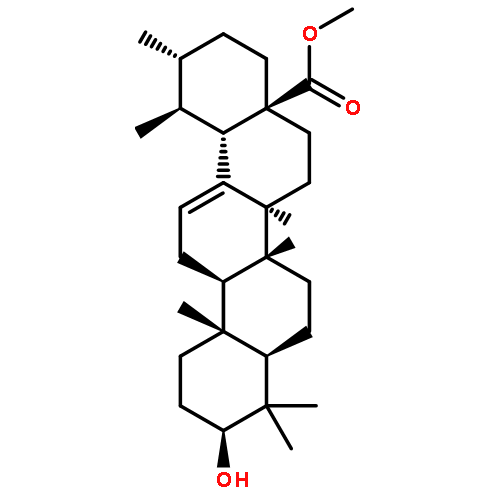
Co-reporter:Anne Loesche, Jana Wiese, Sven Sommerwerk, Vivienne Simon, Wolfgang Brandt, René Csuk
European Journal of Medicinal Chemistry 2017 Volume 125() pp:430-434
Publication Date(Web):5 January 2017
DOI:10.1016/j.ejmech.2016.09.051
•Twenty one analogs of the antiseptic agent picloxydine were synthesized.•They were screened for their ability to inhibit cholinesterases AChE and BChE.•A p-nitro substituted bisbiguanide 43 was a good inhibitor for AChE.•A p-tert-butyl substituted analog 42, however, inhibited BChE in the low μM range.Drug repurposing (=drug repositioning) is an effective way to cut costs for the development of new therapeutics and to reduce the time-to-market time-span. Following this concept a small library of compounds was screened for their ability to act as inhibitors of acetyl- and butyrylcholinesterase. Picloxydine, an established antiseptic, was shown to be an inhibitor for both enzymes. Systematic variation of the aryl substituents led to analogs possessing almost the same good properties as gold standard galantamine hydrobromide.
Co-reporter:Annemarie E. Kramell;Alexander O. Brachmann;Ralph Kluge;Jörn Piel;René Csuk
RSC Advances (2011-Present) 2017 vol. 7(Issue 21) pp:12990-12997
Publication Date(Web):2017/02/21
DOI:10.1039/C6RA27842F
Dyestuff analyses were performed directly from the surface of different bluish and reddish colored historic textile samples by flowprobe™-electrospray ionization-high-resolution mass spectrometry (flowprobe™-ESI-HRMS). This real-time in situ microextraction method allowed rapid, reliable and minimal-destructive analysis without extra and time-consuming sample preparation and required only a minimum amount of valuable archaeological material. As demonstrated for indigo-type and anthraquinone dyes this technique is useful for the analysis of various types of textiles regardless of their fiber matrix, appearance or handicraft and is also suitable for investigating fragile archeological fibers. Thus, flowprobe™-ESI-HRMS is a promising analytical tool for characterizing organic colorants in objects of archaeological interest.
Co-reporter:Jana Wiemann, Julia Karasch, Anne Loesche, Lucie Heller, Wolfgang Brandt, René Csuk
European Journal of Medicinal Chemistry 2017 Volume 139(Volume 139) pp:
Publication Date(Web):20 October 2017
DOI:10.1016/j.ejmech.2017.07.081
•Piperlongumine B is an alkaloid previously only isolated from long pepper in tiny amounts.•A facile synthesis for Piperlongumine B and analogs has been developed.•These compounds were screened for their potential as inhibitors for cholinesterases.•Screening showed several of them as good inhibitors of acetylcholinesterase.Piperlongumine B (19), an alkaloid previously isolated from long pepper (Piper longum) has been synthesized for the first time in a short sequence and in good yield together with 19 analogs. Screening of these compounds in Ellman's assays showed several of them to be good inhibitors of acetylcholinesterase while being less active for butyrylcholinesterase. Activity of the compounds increased with the ring size of the heterocycle, and a maximum of activity was observed for an analog holding 12 methylene groups in the aliphatic side chain. These compounds may be regarded as promising candidates for the development of efficient inhibitors of acetylcholinesterase being useful for the treatment of Alzheimer's disease.Download high-res image (90KB)Download full-size image
Co-reporter:Sven Sommerwerk, Lucie Heller, Christoph Kerzig, Annemarie E. Kramell, René Csuk
European Journal of Medicinal Chemistry 2017 Volume 127(Volume 127) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.ejmech.2016.12.040
•Rhodamine B conjugates of triterpenoic acids were prepared.•Rhodamine B and the parent triterpenoids were not cytotoxic but the conjugates.•The conjugates were cytotoxic for tumor cells with EC50 at nanomolar concentrations.•Staining experiments showed these compounds to act as mitocans.Triterpenoic acids 1–6 exhibited very low or no cytotoxicity at all, but their corresponding 2,3-di-O-acetyl-piperazinyl amides 13–18 showed low EC50 values for several human tumor cell lines. Their cytotoxicity, however, was also high for the non-malignant mouse fibroblasts NIH 3T3. A significant improvement was achieved by preparing the rhodamine B derivatives 19–24. While rhodamine B is not cytotoxic (up to a concentration of 30μM – cut-off of the assay), the triterpenoid piperazine-spacered rhodamine B derivatives were cytotoxic in nano-molar concentration. Compound 24 (a diacetylated maslinic acid derivative) was most toxic for several human tumor cell lines but less toxic for mouse fibroblasts NIH 3T3. Staining and double-staining experiments revealed 24 to act as a mitocan.Download high-res image (183KB)Download full-size image
Co-reporter:Diego Rodríguez-Hernández, Luiz C.A. Barbosa, Antonio J. Demuner, Amalyn Nain-Perez, Sebastião R. Ferreira, Ricardo T. Fujiwara, Raquel M. de Almeida, Lucie Heller, René Csuk
European Journal of Medicinal Chemistry 2017 Volume 140(Volume 140) pp:
Publication Date(Web):10 November 2017
DOI:10.1016/j.ejmech.2017.09.045
•A series of 18 novel bis-triazolylhederagenin derivatives has been synthesized.•Compound 5 is at least 1780 times more selective than commercial antimony drug.•Derivatives 2, 5 and 17 showed IC50 around of 5–29 μM against L. infantum.•Derivative 19 was the most cytotoxic with EC50 around of 7.4–12.1 μM.•Hederagenin and 2, 5, 17 interact in the binding site of the enzyme CYP51Li.Aiming to obtain new potent leishmanicidal and cytotoxic compounds from natural sources, the triterpene hederagenin was converted into several new 1,2,3-triazolyl derivatives tethered at C-23 and C-28. For this work hederagenin was isolated from fruits of Sapindus saponaria and reacted with propargyl bromide to afford as a major product bis-propargylic derivative 1 in 74%. Submitting this compound to Huisgen 1,3-dipolar cycloaddition reactions with several azides afforded the derivatives 2–19 with yields in the range of 40–87%. All compounds have been screened for in vitro cytotoxic activity in a panel of five human cancer cell lines by a SRB assay. The bioassays showed that compound 19 was the most cytotoxic against all human cancer cell lines with EC50 = 7.4–12.1 μM. Moreover, leishmanicidal activity was evaluated through the in vitro effect in the growth of Leishmania infantum, and derivatives 1, 2, 5 and 17 were highly effective preventing proliferation of intracellular amastigote forms of L. infantum (IC50 = 28.8, 25.9, 5.6 and 7.4 μM, respectively). All these compounds showed a higher selectivity index and low toxicity against two strains of kidney BGM and liver HepG2 cells. Compound 5 has higher selectivity (1780 times) in comparison with the commercial antimony drug and is around 8 times more selective than the most active compound previously reported hederagenin derivative. Such high activity associated with low toxicities make the new bis-traiazolyl derivatives promising candidates for the treatment of leishmaniasis. In addition, hederagenin and some derivatives (2, 5 and 17) showed interaction in the binding site of the enzyme CYP51Li.Download high-res image (159KB)Download full-size image
Co-reporter:Lucie Heller, Michael Kahnt, Anne Loesche, Patricia Grabandt, Stefan Schwarz, Wolfgang Brandt, René Csuk
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.11.056
•A set of 35 30-nor-lupane derivatives was prepared from platanic acid.•The compounds were tested for their cholinesterase activity.•One compound was a strong inhibitor for equine butyrylcholinesterase (BChE).•This compound showed a low Ki = 0.01 μM and high selectivity for BChE (FB = 851).A set of thirtyfive 30-norlupan derivatives (2–36) was prepared from the natural triterpenoid platanic acid (PA), and the hydroxyl group at C-3, the carboxyl group at C-17 and the carbonyl group at C-20 were modified. These derivatives were tested for their inhibitory activity for the enzymes acetylcholinesterase (AChE, from electric eel) and butyrylcholinesterase (BChE, from equine serum) using Ellman's assay. Extra enzyme kinetic studies were performed. The most active compound was (3β, 20R)-3-acetyloxy-20-amino-30-norlupan-28-oate (32) showing a Ki value of 0.01 ± 0.003 μM for BChE. This compound proved to be a selective (FB = 851), mixed-type inhibitor for BChE.Download high-res image (159KB)Download full-size image
Co-reporter:Jana Wiemann, Anne Loesche, René Csuk
Bioorganic Chemistry 2017 Volume 74(Volume 74) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.bioorg.2017.07.013
•Dehydroabietylamides have been prepared and characterized.•They were screened for their activity to inhibit acetyl- and butyrylcholinesterase.•Inhibition of butyrylcholinesterase was low.•However, several of them were good to excellent inhibitors of acetylcholinesterase.•N-Benzoyldehydroabietylamides showed inhibition rates comparable to galantamine.Nowadays, the inhibition of acetylcholinesterase is one of the main pharmacological strategies for the treatment of Alzheimer’s disease. Therefore, a set of thirty-four derivatives of the diterpenoid dehydroabietylamine has been synthesized and screened in colorimetric Ellman’s assays to determine their ability to inhibit the enzymes acetylcholinesterase (AChE, from electric eel) and butyrylcholinesterase (BChE, from equine serum). A systematic variation of the substitution of dehydroabietylamides enabled an approach to analogs showing a remarkable inhibition potency for AChE. Particularly N-benzoyldehydroabietylamines 11, 12 and 13 were excellent inhibitors for AChE, showing inhibition rates comparable to standard galantamine hydrobromide.Download high-res image (61KB)Download full-size image
Co-reporter:Diego Rodríguez-Hernández, Antonio J. Demuner, Luiz C.A. Barbosa, Lucie Heller, René Csuk
European Journal of Medicinal Chemistry 2016 Volume 115() pp:257-267
Publication Date(Web):10 June 2016
DOI:10.1016/j.ejmech.2016.03.018
•A series of novel aryl-1H-1,2,3-triazol-4-ylesters and amides derivatives of hederagenin has been synthesized.•Some derivatives were more active for six human cancer lines than hederagenin.•EC50 values for m-Br, m-Cl and m-NO2 ester derivatives ranged 3.0–4.1 μM.•An ester derivative with an o-F group was the most cytotoxic compound against HT29 cells showing EC50 = 1.6 μM and IS = 5.4.A series of novel aryl-1H-1,2,3-triazol-4-yl methylester and amide derivatives of the natural product hederagenin was synthesized aiming to develop new antitumor agents, using Huisgen 1,3-dipolar cycloaddition reactions, with yields between 35% and 95%. The structures of all derivatives (2–31) were confirmed by MS, IR, 1H NMR and 13C NMR spectroscopic data. The cytotoxic activities of all compounds were screened against a panel of six human cancer cell lines using SRB assay. It was found that most of the compounds displayed higher levels of antitumor activities as compared to parent hederagenin. Compounds 4, 8 and 15 were the most potent against all human cancer cell lines. Furthermore, compound 11 was the most cytotoxic against cell HT29 showing EC50 = 1.6 μM and a selectivity index of 5.4.
Co-reporter:Sven Sommerwerk, Lucie Heller, Julia Kuhfs, René Csuk
European Journal of Medicinal Chemistry 2016 Volume 119() pp:1-16
Publication Date(Web):25 August 2016
DOI:10.1016/j.ejmech.2016.04.051
•Urea derivatives of ursolic, oleanolic and maslinic acid were synthesized.•They were tested for their antitumor activity using human cancer cell lines.•A (2β,3β)-diacetoxy moiety in an oleanane skeleton increases cytotoxicity.•The compounds are cytostatic and act by apoptosis.2,3-Di-O-acetyl-maslinic acid benzylamide (5) has previously been shown to possess high cytotoxicity for a variety of human tumor cell lines while being of low cytotoxicity to non-malignant cells. Structural modifications performed on 5 revealed that the presence of these acetyl groups in 5 and the presence of (2β,3β)-configurated centers seems necessary for obtaining high cytotoxicity combined with best selectivity between malignant cells and non-malignant mouse fibroblasts. Compounds carrying an ursane skeleton showed weaker cytotoxicity than their oleanane derived analogs. In addition, the benzylamide function in compound 5 should be replaced by a phenylurea moiety to gain better cytotoxicity while retaining and improving the selectivity. Thus, maslinic acid derived N-[2β,3β-di-O-acetyl-17β-amino-28-norolean-12-en-17-yl]phenylurea (45) gave best results showing EC50 = 0.9 μM (for A2780 ovarian cancer cells) with EC50 > 120 μM for fibroblasts (NIH 3T3) and triggered apoptosis while caspase-3 was not activated by this compound.
Co-reporter:Sven Sommerwerk, Lucie Heller, Julia Kuhfs, René Csuk
European Journal of Medicinal Chemistry 2016 Volume 122() pp:452-464
Publication Date(Web):21 October 2016
DOI:10.1016/j.ejmech.2016.06.053
•Amide derivatives of augustic, 2-epi-corosolic and asiatic acid were synthesized.•They were tested for their antitumor activity using human cancer cell lines.•An augustic acid derived 4-isoquinolinyl amide showed increased cytotoxicity.•A 4-isoquinolinyl derivative of asiatic acid (28) gave EC50 = 80 nM (A2780 cells).•The compounds act by apoptosis.2,3-Di-O-acetyl-triterpenoic acid derived amides possessing a (2β, 3β) configuration in ring A and two acetyl groups were previously shown to possess high cytotoxicity for human tumor cell lines but to exhibit low cytotoxicity for non-malignant mouse fibroblasts. In this study, augustic acid (1) and 2-epi-corosolic acid (2) were chosen as starting points for the synthesis of analogs. While augustic acid derived 3-quinolinyl amide 9 gave low EC50 values in SRB assays but was cytotoxic for all lines, the isomeric 4-isoquinolinyl amide 21 was very cytotoxic for the tumor cell lines but significantly less cytotoxic for the mouse fibroblasts NIH 3T3. In addition, a triacetylated 4-isoquinolinyl derivative of asiatic acid (28) gave EC50 = 80 nM (for A2780 ovarian cancer cells). As shown by additional experiments (acridine orange/propidium iodide staining, fluorescence spectroscopy and cell cycle investigations) these compounds act mainly by apoptosis.A small structural difference (3-quinolinyl/4-isoquinolinyl) has a strong impact on cytotoxicity and malignant/non-malignant cell selectivity.
Co-reporter:Jana Wiemann, Lucie Heller, René Csuk
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 3) pp:907-909
Publication Date(Web):1 February 2016
DOI:10.1016/j.bmcl.2015.12.064
Oleanolic and ursolic acid derived hydroxamates were easily obtained from their parent compounds; they were screened for their cytotoxicity applying SRB assays employing several human tumor cell lines. Low EC50 values were determined for compounds in which the nitrogen as well as the oxygen in the hydroxamic acid part still holds acidic hydrogens. Thus, ursolic acid derived compounds having at least an OH and/or NH moiety in the hydroxamate part of the molecule showed good cytotoxicity but they are significantly less selective for the tumor cells than oleanolic acid derived compounds. Good results were determined for oleanolic acid derived 7 for tumor cell lines 518A2 (melanoma, EC50 = 3.3 μM), A2780 (ovarian carcinoma, EC50 = 3.4 μM) and HT29 (colon adenocarcinoma, EC50 = 5.6 μM) while being significantly less cytotoxic for fibroblasts (EC50 = 20.4 μM).
Co-reporter:Lucie Heller, Anja Knorrscheidt, Franziska Flemming, Jana Wiemann, Sven Sommerwerk, Ioana Z. Pavel, Ahmed Al-Harrasi, René Csuk
Bioorganic Chemistry 2016 Volume 68() pp:137-151
Publication Date(Web):October 2016
DOI:10.1016/j.bioorg.2016.08.004
•3-O-Acetyl-OA derived amides have been prepared.•They exhibited fair to good cytotoxicity for several human tumor cell lines.•Picolinylamides acted by autophagy and arrest of the cell cycle in the S phase.•A N-[2-(dimethylamino)-ethyl] derivative triggered apoptosis.•Small structural differences result great differences cytotoxicity of analogs.Thirty-one different 3-O-acetyl-OA derived amides have been prepared and screened for their cytotoxic activity. In the SRB assays nearly all the carboxamides displayed good cytotoxicity in the low μM range for several human tumor cell lines. Low EC50 values were obtained especially for the picolinylamides 14–16, for a N-[2-(dimethylamino)-ethyl] derivative 27 and a N-[2-(pyrrolinyl)-ethyl] carboxamide 28. These compounds were submitted to an extensive biological testing and proved compound 15 to act mainly by an arrest of the tumor cells in the S phase of the cell cycle. Cell death occurred by autophagy while compounds 27 and 28 triggered apoptosis.
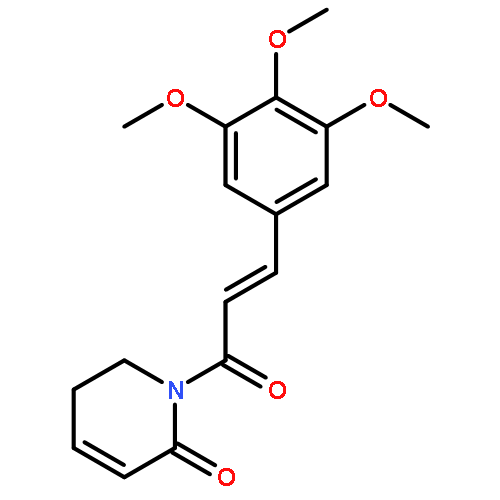
Co-reporter:Sven Sommerwerk, Ralph Kluge, Dieter Ströhl, Lucie Heller, Annemarie Elisabeth Kramell, Sven Ogiolda, Phil Liebing, René Csuk
Tetrahedron 2016 Volume 72(Issue 11) pp:1447-1454
Publication Date(Web):17 March 2016
DOI:10.1016/j.tet.2016.01.045
Several head-to-head and head-to-tail dimers of piplartine (1a) were prepared, and the configurations of the resulting truxillic and truxinic acid derivatives were established by a combination of NMR experiments and single-crystal X-ray analysis. Their cytotoxic activity was screened in photometric sulforhodamine B assays. All of these dimers showed a decreased cytotoxicity compared to 1a except for the β-truxinic acid derivative 3a. For this unprecedented head-to-head dimer high cytotoxic activity was established against several human tumor cell lines, and IC50 values as low as 1.1 μM were found.
Co-reporter:Diego Rodríguez-Hernández, Antonio J. Demuner, Luiz C.A. Barbosa, René Csuk, Lucie Heller
European Journal of Medicinal Chemistry 2015 Volume 105() pp:57-62
Publication Date(Web):13 November 2015
DOI:10.1016/j.ejmech.2015.10.006
•A series of ester and amide derivatives of hederagenin has been synthesized.•Some derivatives were more actives than hederagenin for six human cancer lines.•Amides with pyrimidinyl and pyrrolidinyl groups were the most active derivatives.•EC50 values for pyrimidinyl and pyrrolidinyl derivatives ranged from 1.1 to 6.5 μM.•AO/PI staining experiment showed that compound 28 mainly acts by apoptosis.In this study, a series of novel C-28 esters and amides derivatives of hederagenin (He) were designed and synthesized in attempt to develop potent antitumor agents. Their structures were confirmed by MS, IR, 1H NMR and 13C NMR spectroscopic analyses and their cytotoxic activities were screened in SRB assays using a panel of six human cancer cell lines. Although most of the compounds displayed moderate to high levels of cytotoxic activity they were all more potent than the natural product He. The most active compounds had either an ethylpyrimidinyl (27) or an ethylpyrrolidinyl (28) substituent, with EC50 in the range of 1.1–6.5 μM for six human cancer cell lines. Notably, this corresponds to an approximately 30-fold times greater potency than He.
Co-reporter:Stefan Schwarz, Anne Loesche, Susana Dias Lucas, Sven Sommerwerk, Immo Serbian, Bianka Siewert, Elke Pianowski, René Csuk
European Journal of Medicinal Chemistry 2015 Volume 103() pp:438-445
Publication Date(Web):20 October 2015
DOI:10.1016/j.ejmech.2015.09.007
•In this study we investigated a variety of derivatives of maslinic acid.•Maslinic acid could be converted into a selective inhibitor for acetylcholinesterase.•The compounds are no substrates for other hydrolytic enzymes.•The compounds show rather low cytotoxicity.•Their activity is similar to that of standard compound galantamine.During the last decade, maslinic acid has been evaluated for many biological properties, e.g. as an anti-tumor or an anti-viral agent but also as a nutraceutical. The potential of maslinic acid and related derivatives to act as inhibitors of acetyl- or butyryl-cholinesterase was examined in this communication in more detail. Cholinesterases do still represent an interesting group of target enzymes with respect to the investigation and treatment of the Alzheimer's disease and other dementia illnesses as well. Although other triterpenoic acids have successfully been tested for their ability to act as inhibitors of cholinesterases, up to now maslinic acid has not been part of such studies.For this reason, three series of maslinic acid derivatives possessing modifications at different centers were synthesized and subjected to Ellman's assay to determine their inhibitory strength and type of inhibitory action. While parent compound maslinic acid was no inhibitor in these assays, some of the compounds exhibited an inhibition of acetylcholinesterase in the single-digit micro-molar range. Two compounds were identified as inhibitors of butyrylcholinesterase showing inhibition constants comparable to those of galantamine, a drug often used in the treatment of Alzheimer's disease. Furthermore, additional selectivity as well as cytotoxicity studies were performed underlining the potential of several derivatives and qualifying them for further investigations. Docking studies revealed that the different kinetic behavior within the same compound series may be explained by the ability of the compounds to enter the active site gorge of AChE.
Co-reporter:René Csuk, Anja Niesen-Barthel, Renate Schäfer, Alexander Barthel, Ahmed Al-Harrasi
European Journal of Medicinal Chemistry 2015 Volume 92() pp:700-711
Publication Date(Web):6 March 2015
DOI:10.1016/j.ejmech.2015.01.039
•We investigated a variety of derivatives of 11-keto-β-boswellic acid.•The compounds showed antitumor activity on different human cancer cell lines.•Many of the compounds are cytotoxic even in a low micro mol concentration.•A nitrogen bearing substituent at position C-2 increases cytotoxicity.•Introduction of an additional carboxylic group decreases cytotoxicity.Beta-boswellic acids are interesting triterpenoic acids that show different biological activities. Their cytotoxic potential, as well as that of their derivates remained unexploited so far. In this study we were able to prepare derivatives of 11-keto-β-boswellic acid that showed lower IC50 values as determined by a sulphorhodamine B (SRB) assay using several different human tumour cell lines. Thus, the introduction of an amino group at position C-2 led to a significantly improved cytotoxic activity of amine 18. An apoptotic effect of compound 18 was determined using DNA laddering and trypan blue staining experiments.
Co-reporter:Lucie Heller;Sven Sommerwerk;Felix Tzschöckell;Jana Wiemann;Stefan Schwarz;Bianka Siewert;Ahmed Al-Harrasi;René Csuk
Archiv der Pharmazie 2015 Volume 348( Issue 12) pp:889-896
Publication Date(Web):
DOI:10.1002/ardp.201500318
(18α)-Glycyrrhetinic acid (4) was prepared from (18β)-glycyrrhetinic acid (1), and the cytotoxicity of some derivatives was investigated by photometric SRB assays employing several human tumor cell lines. In summary, (18β)-1 is slightly more cytotoxic than its (18α) epimer 4, but its cytotoxicity is negligible. Higher cytotoxicity was observed for the esters 2 and 5 and for the 3-O-acetylated esters 3 and 6. Cytotoxicity was improved dramatically when the hydroxyl group at position C-3 was replaced by an amino moiety. SeO2 oxidations gave access to a novel furano-glycyrrhetinoate 15. Interestingly, its seleno analog 16 is approximately five to six times less cytotoxic for the tumor cell lines tested, and tumor/non-tumor selectivity is lost upon replacement of the oxygen by a selenium substituent.
Co-reporter:Sven Sommerwerk;Lucie Heller ;René Csuk
Archiv der Pharmazie 2015 Volume 348( Issue 1) pp:46-54
Publication Date(Web):
DOI:10.1002/ardp.201400297
Methyl triterpenoates derived from oleanolic, ursolic, betulinic, glycyrrhetinic, platanic, or maslinic acid were converted into their corresponding sulfamates and carbamoylsulfamates. The sulfamates were screened in photometric sulforhodamine assays for cytotoxic activity employing several human tumor cell lines. Many of the compounds showed EC50 values in one-digit μM concentration. Of special interest seems methyl (3β) 3-(aminosulfonyloxy)-11-oxo-oleanoate (18) showing good cytotoxicity for the human adenocarcinomic alveolar basal epithelial cell line A549 while being less toxic for non-malignant NIH 3T3 mouse fibroblasts.
Co-reporter:Lucie Heller, Anja Obernauer, René Csuk
Bioorganic & Medicinal Chemistry 2015 23(13) pp: 3002-3012
Publication Date(Web):
DOI:10.1016/j.bmc.2015.05.015
Co-reporter:Sven Sommerwerk, Lucie Heller, Immo Serbian, René Csuk
Tetrahedron 2015 Volume 71(Issue 45) pp:8528-8534
Publication Date(Web):11 November 2015
DOI:10.1016/j.tet.2015.09.037
The four diastereomeric 2,3-dihydroxy-olean-12-en-28-oic acids (maslinic, augustic, bredemolic and 3-epi-maslinic acid) were easily accessed from one single starting material, oleanolic acid. The procedures allow the medium-to-large scale preparation of these valuable starting materials. Except for maslinic acid, the triterpenoic acids showed only a low cytotoxicity towards several human tumor cell lines.
Co-reporter:Lucie Heller, Stefan Schwarz, Vincent Perl, Alexander Köwitsch, Bianka Siewert, René Csuk
European Journal of Medicinal Chemistry 2015 101() pp: 391-399
Publication Date(Web):
DOI:10.1016/j.ejmech.2015.07.004
Co-reporter:Bianka Siewert, René Csuk
European Journal of Medicinal Chemistry 2014 Volume 74() pp:1-6
Publication Date(Web):3 March 2014
DOI:10.1016/j.ejmech.2013.12.031
•A derivative of maslinic acid is incorporated into tumor cell's membrane.•Cholesterol from the membrane is extruded.•This damaging of the cell membrane initiates apoptosis.•Co-crystals consisting of this analog and cholesterol are formed.•This extends previous models on apoptosis initiating mechanisms.Close inspection of human ovarian cancer cells A2780 in the course of an antitumor screening using maslinic acid analogs revealed for one of the compounds, 4-oxa-4-phenyl-butyl 2,3-dihydroxy-olean-12-en-28-oate (1), an unusual behavior. During the incubation of the cells with 1, at the perimeter of the cells or close by crystals were formed consisting of cholesterol and excess 1. Compound 1 was incorporated into the cell's membrane followed by an extrusion of cholesterol from the lipid rafts. As a consequence of the alterations of the cell membrane, a volume decrease was initiated that triggered apoptosis; this extends previous models on apoptosis initiating mechanisms.Incubation of human ovarian cancer cells A2780 with 4-oxa-4-phenyl-butyl 2,3-dihydroxy-olean-12-en-28-oate (1) resulted in an incorporation of 1 into the cell's membrane followed by an extrusion of cholesterol from the lipid rafts.
Co-reporter:Stefan Schwarz, Bianka Siewert, Nuno M. Xavier, Ana R. Jesus, Amélia P. Rauter, René Csuk
European Journal of Medicinal Chemistry 2014 Volume 72() pp:78-83
Publication Date(Web):24 January 2014
DOI:10.1016/j.ejmech.2013.11.024
•In this study we synthesized and investigated monodesmosidic glycyrrhetinic acid glycosides.•The compounds were tested for their antitumor activity using human cancer cell lines.•Many of the compounds are cytotoxic.•The compounds act by apoptosis.Several pentacyclic triterpenoic acids have shown noteworthy antitumor activity, among them betulinic acid as well as oleanolic acid and derivatives thereof. Glycyrrhetinic acid (GA) exhibits some cytotoxic activity albeit this compound is not as active as betulinic acid, but GA came in the focus of scientific interest since it triggers apoptosis in tumor cells. In addition, it can be extracted from the roots of liquorice in high yields. Previous studies revealed that the introduction of an extra hydrophilic moiety increases the cytotoxicity of these compounds. Thus, a series of GA glycosides was prepared utilizing hexoses as well as pentoses (in d- and l-configuration) by using glycosyl trichloroacetimidates and TMSOTf as catalyst. The compounds were screened for cytotoxic activity against seven human cancer cell lines and the not malignant murine cell line NIH 3T3using a photometric SRB assay. The compounds trigger apoptosis as shown from extra trypan blue and acridine orange/ethidium bromide staining.Several novel monodesmosidic derivatives of methyl glycyrrhetinate were prepared. They were shown to be cytotoxic against several human tumor cell lines and to trigger apoptotic cell death.
Co-reporter:Bianka Siewert, Jana Wiemann, Alexander Köwitsch, René Csuk
European Journal of Medicinal Chemistry 2014 Volume 72() pp:84-101
Publication Date(Web):24 January 2014
DOI:10.1016/j.ejmech.2013.11.025
•C ring modified oleanolic, ursolic and maslinic acid derivatives were synthesized.•The compounds showed antitumor activity using different human cancer cell lines.•Many of the compounds are cytotoxic even in a low μmol concentration.•The compounds act by apoptosis.•Small changes in constitution cause tremendous effects in their mode of action.A convenient and elegant route has been developed to separate the natural regioisomers triterpenoids ursolic acid (UA) and oleanolic acid (OA) as well as derivatives thereof. Eleven unknown derivatives of OA were designed, synthesized, and their cytotoxicity was investigated. Further sixteen compounds were prepared to correlate all compounds in a SAR study. It could be shown that C-ring modifications of OA and UA have only a moderate influence onto the cytotoxic activity of the compounds but a significant impact onto the ability to trigger apoptosis in ovarian cancer cells (cell line A2780).C-ring modifications of oleanolic and ursolic acid have only a moderate influence onto the cytotoxic activity of the compounds but the intact presence of this structural element has a significant impact onto the ability to trigger apoptosis in ovarian cancer cells (cell line A2780).
Co-reporter:Stefan Schwarz, Sven Sommerwerk, Susana D. Lucas, Lucie Heller, René Csuk
European Journal of Medicinal Chemistry 2014 Volume 86() pp:95-102
Publication Date(Web):30 October 2014
DOI:10.1016/j.ejmech.2014.08.051
•Sulfamates of methyl triterpenoates were synthesized.•Several compounds were inhibitors for carbonic anhydrase II.•They act as competitive enzyme inhibitors even in a low μmol concentration.•Small changes in constitution cause tremendous effects in their activity.Carbonic anhydrase II, belonging to one of the most important enzyme groups of the human body, is a well-studied isozyme from the family of the carbonic anhydrases. Since it is involved in several physiological processes, it has been a pharmaceutical target for many years. In this study we synthesized a number of sulfamates derived from pentacyclic methyl triterpenoates, and we demonstrate their potential as carbonic anhydrase II inhibitors using the well-established photometric 4-nitrophenyl acetate assay. Inhibition constants, as an indicator of their inhibition strength, were in the micromolar range; one compound (10, methyl (3β) 3-(aminosulfonyloxy)-oleanoate) showed a Ki value as low as 0.3 μM. This Ki value is comparable to that of acetazolamide which is a potent carbonic anhydrase inhibitor and a drug for the treatment of glaucoma.
Co-reporter:Bianka Siewert, Elke Pianowski, Anja Obernauer, René Csuk
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 1) pp:594-615
Publication Date(Web):1 January 2014
DOI:10.1016/j.bmc.2013.10.047
Several novel esters and amides of maslinic acid were prepared. Their evaluation for cytotoxic activity with a panel of human cancer cell lines using a sulforhodamine B (SRB) assay revealed for some of them a noteworthy activity. The results from annexinV-FITC and caspase-assays as well as from DNA laddering experiments provided evidence for an apoptotic cell death. A diacetylated benzylamide (15) induced a G1/G0 arrest in tumor cells. It also displayed an extraordinary cytotoxicity against human ovarian cancer cells but a 300 times lower toxicity for non-malignant primary human fibroblasts.
Co-reporter:René Csuk, Lucie Heller, Bianka Siewert, Andrey Gutnov, Oliver Seidelmann, Volkmar Wendisch
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 16) pp:4011-4013
Publication Date(Web):15 August 2014
DOI:10.1016/j.bmcl.2014.06.021
β-Nitro-substituted ethyl carboxylates are a new class of cytotoxic agents; they can be easily obtained in fair to good yields in a single-step reaction by a Pd-catalyzed asymmetric conjugate addition of aryl boronic acids to 2-nitro-acrylates. Of all the tested derivatives, 2-(4-chlorophenyl)-3-nitropropionic acid ethyl ester (6) is most cytotoxic especially against the human ovarian cancer cell line A2780 therefore making this compound an interesting candidate for further investigations.
Co-reporter:Sven Sommerwerk, René Csuk
Tetrahedron Letters 2014 Volume 55(Issue 37) pp:5156-5158
Publication Date(Web):10 September 2014
DOI:10.1016/j.tetlet.2014.07.074
•A convenient synthesis of triterpenoid maslinic acid has been developed.•The synthesis of maslinic acid is very short and provides high yields.•The synthesis is totally chromatography-free.•The procedure can be scaled-up very easily.•Slight modifications allow the synthesis of isomeric augustic acid.A convenient and chromatography-free 4-step synthesis of analytically pure maslinic acid (1, 41.2%) from oleanolic acid has been developed. Slight variations in the final steps gave an excellent yield of isomeric augustic acid (7, 71.9%).A convenient and chromatography-free route has been developed for a partial synthesis of maslinic acid and augustic acid from oleanolic acid.
Co-reporter:Sven Sommerwerk, Simone Kern, Lucie Heller, René Csuk
Tetrahedron Letters 2014 Volume 55(Issue 45) pp:6243-6244
Publication Date(Web):5 November 2014
DOI:10.1016/j.tetlet.2014.09.079
•A convenient synthesis of piperodione has been developed.•The synthesis of piperodione is very short and provides high yields.•Key steps of the synthesis are a Mannich and a Stetter reaction.•The route allows the synthesis of analogs.•Piperodione as well as analogs show no cytotoxicity.Piperodione (3), a secondary metabolite previously isolated from the Javanese pepper plant Piper retrofractum in 0.0002% isolated yield was synthesized in a convergent strategy utilizing a Mannich and a Stetter reaction. An over-all yield of 76% could be achieved. Several analogs were prepared by this synthetic sequence. None of the compounds showed a significant cytotoxicity for human tumor cells (photometric sulforhodamine B assay).
Co-reporter:Lucie Heller;Stefan Schwarz;Björn A. Weber ;René Csuk
Archiv der Pharmazie 2014 Volume 347( Issue 10) pp:707-716
Publication Date(Web):
DOI:10.1002/ardp.201400103
Gypsogenin (1) was obtained by acidic hydrolysis from its saponin. While the parent compound 1 acted as a selective inhibitor for butyrylcholinesterase (from equus) possessing a moderate mixed-type inhibition of the enzyme, Ki values as low as 2.67 ± 0.59 μM were determined for (3β,4α) 3-O-acetyl-olean-12-ene-23,28-dinitrile (11) and acetylcholinesterase (AChE, from electric eel). Thus, 11 possesses one-fifth of the inhibitory activity of the “gold standard” galantamine hydrobromide; this compound is one of the first pentacyclic triterpenoids described as a potent AChE-selective inhibitor.
Co-reporter:Stefan Schwarz, Susana Dias Lucas, Sven Sommerwerk, René Csuk
Bioorganic & Medicinal Chemistry 2014 22(13) pp: 3370-3378
Publication Date(Web):
DOI:10.1016/j.bmc.2014.04.046
Co-reporter:René Csuk, Ronny Sczepek, Bianka Siewert, Christoph Nitsche
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 2) pp:425-435
Publication Date(Web):15 January 2013
DOI:10.1016/j.bmc.2012.11.016
Several novel betulin derivatives were prepared and evaluated for their antitumor activity. Among others, 3-O-acetylbetulinic aldehyde served as an ideal starting material for the synthesis of 28-acetylenic derivatives that were further transformed into Mannich bases. These hydroxypropargylamines were screened for their antitumor activity in a panel of nine human cancer cell lines in a sulforhodamine B (SRB) assay. Several compounds showed a noteworthy antitumor activity. The results from acridine orange/propidium iodide staining and annexinV-FITC assays as well as DNA laddering experiments provided evidence for an apoptotic cell death.
Co-reporter:René Csuk, Bianka Siewert, Jana Wiemann
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 12) pp:3542-3546
Publication Date(Web):15 June 2013
DOI:10.1016/j.bmcl.2013.04.036
During the reaction of methyl 3β-acetoxy-glycyrrhetinate (1) with SeO2 significant amounts of a cytotoxic hitherto unprecedented triterpenoic selenophene 3 are formed. This compound stops cell proliferation and acts by apoptosis.
Co-reporter:René Csuk, Andrea Schultheiß, Sven Sommerwerk, Ralph Kluge
Tetrahedron Letters 2013 Volume 54(Issue 18) pp:2274-2276
Publication Date(Web):1 May 2013
DOI:10.1016/j.tetlet.2013.02.076
Co-reporter:René Csuk;Christoph Nitsche;Ronny Sczepek;Stefan Schwarz ;Bianka Siewert
Archiv der Pharmazie 2013 Volume 346( Issue 3) pp:232-246
Publication Date(Web):
DOI:10.1002/ardp.201200428
Abstract
Several novel betulin derivates were prepared using Mannich reactions as a key step. Starting from 3-ethynyl-3-hydroxy-lup-20(29)-ene derivatives, copper-catalyzed Mannich reactions yielded hydroxypropargyl ammonium hydrochlorides or their corresponding methiodides. All compounds were screened in a sulforhodamine B assay for their antitumor activity using a panel of 9 human cancer cell lines. Some of these compounds showed significant cytotoxicity; they act by triggering apoptotic cell death as shown by additional acridine orange/propidium iodide assays, Trypan blue tests, DNA laddering experiments, and investigations of the cell cycle.
Co-reporter:René Csuk;Sabrina Albert;Ralph Kluge ;Dieter Ströhl
Archiv der Pharmazie 2013 Volume 346( Issue 7) pp:499-503
Publication Date(Web):
DOI:10.1002/ardp.201300051
Novel polyhydroxylated (E)-stilbenes were synthesized by Mizoroki–Heck reactions and tested for their ability to inhibit the enzymes acetyl- and butyrylcholinesterase. Several of them are good inhibitors of butyrylcholinesterase; one of them carrying an extra fluorine substituent is a 94-fold stronger inhibitor of butyrylcholinesterase than of acetylcholinesterase.
Co-reporter:René Csuk;Stefan Schwarz;Ralph Kluge ;Dieter Ströhl
Archiv der Pharmazie 2012 Volume 345( Issue 1) pp:28-32
Publication Date(Web):
DOI:10.1002/ardp.201000327
Abstract
Several triterpenoic acids display a remarkable cytotoxicity on tumor cells. Glycyrrhetinic acid – the main content of the licorice root – possesses an apoptotic effect on tumor cells. Previous studies pointed out the presence of a keto group at position C-11 in glycyrrhetinic acid derivatives as the main reason for its apoptotic activity. Several pairs of derivatives were synthesized differing only at position C-11. These compounds were tested in a sulforhodamine B colorimetric assay for cytotoxicity screening on 12 tumor cell lines and mouse embryonic fibroblasts (NIH3T3). Our results show that there is no direct relation between the existence of the C-11 keto group and the apoptotic activity of the compounds.
Co-reporter:René Csuk;Stefan Schwarz;Bianka Siewert;Ralph Kluge ;Dieter Ströhl
Archiv der Pharmazie 2012 Volume 345( Issue 3) pp:223-230
Publication Date(Web):
DOI:10.1002/ardp.201100046
Abstract
The extracts of the roots of licorice have been used in traditional and folk medicine to treat a broad variety of maladies. The main ingredient of these extracts is glycyrrhicinic acid. Its aglycon, glycyrrhetinic acid, has many biological activities, among them a pronounced cytotoxicity against tumor cells. In this study we varied glycyrrhetinic acid at position C-30 to get “simple” derivatives, for example esters, amides and a nitrile. The influence of these changes on the cytotoxic activity is noteworthy and was determined by a colorimetric sulphorhodamine B test using 7 human tumor cell lines and mouse embryonic fibroblasts (NIH3T3) for comparison. A Trypan blue test as well as an acridine orange/ethidium bromide test was used to discover the ability of the compounds to induce apoptosis.
Co-reporter:René Csuk;Anke Heinold;Bianka Siewert;Stefan Schwarz;Alexer Barthel;Ralph Kluge ;Dieter Ströhl
Archiv der Pharmazie 2012 Volume 345( Issue 3) pp:215-222
Publication Date(Web):
DOI:10.1002/ardp.201100065
Abstract
Arglabin derivatives varied at the endo- or exo-cyclic double bond were synthesized and studied in a colorimetric sulforhodamine B assay for their cytotoxicity. Variations on the endocyclic double bond led to compounds of reduced cytotoxicity whereas derivatives from the reaction of the α-methylene-γ-butyrolactone moiety led to compounds of similar or only slightly reduced cytotoxicity but different, cell line-dependent selectivity. In addition, arglabin is an excellent starting material for the synthesis of the guaianolide arborescin.
Co-reporter:René Csuk, Stefan Schwarz, Bianka Siewert, Ralph Kluge, Dieter Ströhl
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 11) pp:5356-5369
Publication Date(Web):November 2011
DOI:10.1016/j.ejmech.2011.08.038
Triterpenoic acids show many pharmacological effects, among them an antiinflammatory or an antitumor activity. One of these, glycyrrhetinic acid (1) is of interest because of its antitumor profile. Glycyrrhetinic acid is not only cytotoxic but also triggers apoptosis in various human tumor cell lines. To improve the cytotoxicity of parent 1 we set out to synthesize new derivatives of it – differing in structure and lipophilicity. These compounds were tested in a sulforhodamine B assay for cytotoxicity, and screened for their ability to induce apoptosis using an acridine orange/ethidium bromide assay and trypan blue staining. The most active compound, 34, a benzyl glycyrrhetinate holding an extra 3-N-(3-aminopropyl)glycyl substituent showed IC50 between 1.96 and 5.14 μm for five human cancer cell lines and triggers apoptosis in 80% of the cells.Glycyrrhetinic acid was modified at ring A in many ways. The most active compound, 34, showed excellent IC50 values for five human cancer cell lines and triggers apoptosis.Highlights► In this study we synthesize glycyrrhetinic acid derivatives. ► The compounds were tested for their antitumor activity using human cancer cell lines. ► The derivatives show a significant higher activity than parent glycyrrhetinic acid when attaching an amino group to ring A. ► The compounds act by apoptosis.
Co-reporter:Sabrina Albert, Ralf Horbach, Holger B. Deising, Bianka Siewert, René Csuk
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 17) pp:5155-5166
Publication Date(Web):1 September 2011
DOI:10.1016/j.bmc.2011.07.015
Plants use multiple defence mechanisms comprising both constitutive and inducible barriers to prevent entering of phytopathogenic micro-organisms. In many plant species one of the most efficient responses to combat attacking microbes is the rapid synthesis of antimicrobial low molecular weight phytoalexins, for example, resveratrol, 3,5,4′-trihydroxystilbene (1). Resveratrol and its natural derivatives, however, display only moderate antimicrobial effects. Nevertheless, resveratrol may be a useful lead structure for the chemical synthesis of antimicrobials. In this study, several series of stilbenes have been synthesized, starting from the aldehydes using Wittig reactions to access the corresponding styrenes that were subjected to Mizoroki–Heck reactions to yield the stilbenes in good yields. The stilbenes were tested in an agar diffusion assay against several bacteria and fungi. For some of these compounds the inhibiting zones for bacteria and fungi were comparable with those of the antibiotics tetracycline, streptomycin, ampicillin, or kanamycin, directed against prokaryotes, and nourseothricin or hygromycin controlling fungi, respectively.
Co-reporter:René Csuk, Bianka Siewert
Tetrahedron Letters 2011 Volume 52(Issue 49) pp:6616-6618
Publication Date(Web):7 December 2011
DOI:10.1016/j.tetlet.2011.09.142
A convenient route has been developed to separate regioisomeric ursolic and oleanolic acid by treating the mixture with mCPBA or formic acid/hydrogen peroxide.A convenient route has been developed to separate regioisomeric ursolic and oleanolic acid.
Co-reporter:René Csuk;Alexer Barthel;Ronny Sczepek;Bianka Siewert ;Stefan Schwarz
Archiv der Pharmazie 2011 Volume 344( Issue 1) pp:37-49
Publication Date(Web):
DOI:10.1002/ardp.201000232
Abstract
Novel betulin derivatives were prepared and tested for their antitumor activity. Starting from 3-O-acetyl- or 3-O-methyl-betulinic aldehyde, the synthesis of C-28 ethynyl derivatives was performed; their subsequent transformation with several 1,3-dipolarophiles afforded pyrazoles and 1,2,3-triazoles. Their screening for antitumor activity was performed in a panel of 15 human cancer cell lines by a colorimetric SRB-assay. Thereby, several compounds revealed a higher cytotoxicity than betulinic acid. In addition, the encapsulation of the lead structure 7 into liposomes was investigated. The results from a dye exclusion test and from DNA laddering experiments provided evidence for an apoptotic cell death.
Co-reporter:René Csuk;Stefan Schwarz;Ralph Kluge ;Dieter Ströhl
Archiv der Pharmazie 2011 Volume 344( Issue 8) pp:505-513
Publication Date(Web):
DOI:10.1002/ardp.201100030
Abstract
Glycyrrhetinic acid (GA) is a major ingredient of the dried extract of licorice roots; its antitumor activity is low compared to other members of the triterpenoic family. For example, oleanolic acid, betulin or betulinic acid are more cytotoxic with a pronounced activity for tumor cells. GA, however, is easily to earn, cheap and shows apoptotic effects on tumor cells – like the other triterpenoic acids. These facts bring GA and derivatives in the focus of our scientific interest. Here we tried to improve the poor cytotoxicity of GA by simple derivatization. Thus, we selected various glutamyl and aspartyl substituents for the synthesis of C(3) esters of GA methyl ester. A short (3-5 steps) synthesis was elaborated that allowed to access more effective compounds. One compound, methyl 3β 3-(O-benzyl-L-glutamyl)-11-oxo-olean-12-en-30-oate (5), having a glutamyl substituent with a benzyl protected side chain showed up to 67-fold higher cytoxicity and an up to 140-fold better selectivity towards tumor cells than parent GA. All compounds were evaluated by a sulforhodamine B assay as well as by a trypan blue test and extra acridine orange/ethidium bromide tests for apoptosis.
Co-reporter:René Csuk, Anja Niesen-Barthel, Alexander Barthel, Ralph Kluge, Dieter Ströhl
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 9) pp:3840-3843
Publication Date(Web):September 2010
DOI:10.1016/j.ejmech.2010.05.036
An endoperoxide was synthesized starting from 11-keto-β-boswellic acid and screened for antitumor activity in a panel of 15 human cancer cell lines by an SRB assay. The compound induces apoptosis and shows an average IC50 value of 0.4–4.5 μM.
Co-reporter:René Csuk, Stefan Schwarz, Ralph Kluge, Dieter Ströhl
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 12) pp:5718-5723
Publication Date(Web):December 2010
DOI:10.1016/j.ejmech.2010.09.028
Aminoalkyl substituted derivatives were synthesized starting from glycyrrhetinic acid methyl ester and screened for antitumor activity in a panel of 15 human cancer cell lines by an SRB assay. The most compound 7 possesses an aminohexyl side chain, induces apoptosis and shows IC50 values of 0.6–3.0 μM.
Co-reporter:Stefan Schwarz, René Csuk
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 21) pp:7458-7474
Publication Date(Web):1 November 2010
DOI:10.1016/j.bmc.2010.08.054
Glycyrrhetinic acid (GA) is one of many interesting triterpenoic acids showing anticancerogenic potential. GA is known to trigger apoptosis in tumour cell lines, although GA has a low cytotoxicity. In our study we were able to prepare derivatives of GA that show lowered the IC50 values as determined by a sulforhodamine B (SRB) assay using 15 different human tumour cell lines. Thus, combining an ester group combined with the presence of an amino acid moiety led to a ca. 60-fold improved antitumor activity. Experiments on mouse embryonic fibroblasts (NiH3T3) revealed that these compounds showed a better selectivity for tumour cells compared to the parent compound GA. An apoptotic effect of some of these compounds was determined using an acridine orange/ethidium bromide (AO/EB) test and DNA laddering experiments.
Co-reporter:René Csuk, Alexander Barthel, Ralph Kluge, Dieter Ströhl, Harish Kommera, Reinhard Paschke
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 3) pp:1344-1355
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmc.2009.12.024
The reaction of betulinic aldehydes with various carbon nucleophiles gave a series of new betulin derivatives, among them epoxides, glycidic derivatives and β-hydroxy carbonyl compounds. Subsequent transformations of the β-hydroxy carbonyls lead to 1,3-diketo- and α,β-unsaturated betulin derivatives. These compounds were assayed for cytotoxicity using 15 human cancer cell lines and a colorimetric SRB-assay. Several compounds revealed significant antitumour activity.
Co-reporter:René Csuk, Alexander Barthel, Stefan Schwarz, Harish Kommera, Reinhard Paschke
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 7) pp:2549-2558
Publication Date(Web):1 April 2010
DOI:10.1016/j.bmc.2010.02.042
The plant triterpenes betulin and betulinic acid (BA) are triterpenes featuring interesting pharmacological properties. Starting from substituted betulinic aldehydes, we used them as lead structures for the synthesis of several γ-butyrolactones and butenolides. Their antitumor activity was examined for 15 cancer cell lines using a SRB-assay and their apoptotic action was documented by trypan-blue test and DNA laddering. Several compounds revealed a higher activity than betulinic acid.
Co-reporter:René Csuk, Alexander Barthel, Ralph Kluge, Dieter Ströhl
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 20) pp:7252-7259
Publication Date(Web):15 October 2010
DOI:10.1016/j.bmc.2010.08.023
Several novel betulin derivatives were prepared and evaluated for their antitumor activity. 3-O-Acetylbetulinic aldehyde served as an ideal starting material for the synthesis of 28-acetylenic derivatives. These compounds were further transformed into pyrazoles and 1,2,3-triazoles. Also, the synthesis of 3-amino substituted butenolides was carried out. The compounds were screened for their antitumor activity in a panel of 15 human cancer cell lines in a sulforhodamine B (SRB) assay. Several compounds showed a noteworthy antitumor activity. In addition, the possibility of encapsulation into liposomes was examined, thereby resulting in an increased cytotoxicity. The results from a trypan-blue test and from DNA laddering provided evidence for an apoptotic cell death.
Co-reporter:René Csuk;Erik Prell
Archiv der Pharmazie 2010 Volume 343( Issue 10) pp:577-582
Publication Date(Web):
DOI:10.1002/ardp.200900307
Abstract
Selective difluorination, introducing a lactame moiety (instead of an amine) and a double bond in a trihydroxy-2-thiaquinolizidine derivative reverses the selectivity of the glycosidase inhibitor – a selective inhibitor for an α-glucosidase is altered into an excellent, competitive inhibitor for a β-galactosidase.
Co-reporter:Erik Prell;Claudia Korb;Ralph Kluge;Dieter Ströhl ;René Csuk
Archiv der Pharmazie 2010 Volume 343( Issue 10) pp:583-589
Publication Date(Web):
DOI:10.1002/ardp.200900256
Abstract
Selective monofluorination of the α-glycosidase inhibitor and antidiabetic agent miglitol at positions C(2′) or C(6) creates competitive inhibitors of glycosidases. Introducing a fluorine substituent at position C(6) results in a reduced binding to the enzyme whereas fluorination at C(2′) produces an inhibitor with an activity four times higher than the parent compound. This compound is selective for the α-galactosidase from green coffee beans. Its screening against a panel of human cell lines showed a low cytotoxicity, therefore, making this compound an interesting candidate for further clinical investigations.
Co-reporter:René Csuk, Erik Prell
Tetrahedron 2010 66(6) pp: 1313-1318
Publication Date(Web):
DOI:10.1016/j.tet.2009.12.010
Co-reporter:René Csuk, Erik Prell, Claudia Korb, Ralph Kluge, Dieter Ströhl
Tetrahedron 2010 66(2) pp: 467-472
Publication Date(Web):
DOI:10.1016/j.tet.2009.11.069
Co-reporter:Alexer Barthel;Lothar Trieschmann;Dieter Ströhl;Ralph Kluge;Gerald Böhm;René Csuk
Archiv der Pharmazie 2009 Volume 342( Issue 8) pp:445-452
Publication Date(Web):
DOI:10.1002/ardp.200800196
Abstract
The synthesis of dimeric compounds derived from quinazolin-2-one and 1,4-benzodiazepin-2-one possessing a piperazine or homopiperazine spacer was investigated. In addition, a piperazine spacered bis-isoalloxazine and a bis-riboflavin compound were prepared and their ability to interrupt the association of prion proteins and Alzheimer-specific Aβ peptides was investigated using a fast screening system based on flow cytometry. The bis-isoalloxazine 14 was identified as a new lead structure.
Co-reporter:Anja Niesen;Alexer Barthel;Ralph Kluge;Alexer Köwitzsch;Dieter Ströhl;Stefan Schwarz ;René Csuk
Archiv der Pharmazie 2009 Volume 342( Issue 10) pp:569-576
Publication Date(Web):
DOI:10.1002/ardp.200900051
Abstract
A series of triterpene endoperoxides was synthesized and screened for antitumor activity in a panel of 15 human cancer cell lines by a sulforhodamine-B (SRB) assay. The compounds induce apoptosis and show excellent antitumor activity.
Co-reporter:René Csuk;Alexer Barthel;Christian Raschke;Ralph Kluge;Dieter Ströhl;Lothar Trieschmann;Gerald Böhm
Archiv der Pharmazie 2009 Volume 342( Issue 12) pp:699-709
Publication Date(Web):
DOI:10.1002/ardp.200900065
Abstract
Starting from substituted 9-chloroacridines, a series of quinacrine and spacered dimeric acridine compounds was prepared. Their ability to interrupt the protein association of prion- and Alzheimer-specific proteins and Ab peptides was explored using a fast screening system based on FACS analysis. The bis-acridines displayed a higher activity than the corresponding monomers. Among these derivatives, best results were obtained with the 2,4-dimethoxy-6-nitro compound 7h for Aβ-peptides and the 2-methoxy-6-nitro compound 7f for PrP.
Co-reporter:Erik Prell, René Csuk
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 19) pp:5673-5674
Publication Date(Web):1 October 2009
DOI:10.1016/j.bmcl.2009.08.012
Selective monofluorination of the α-glycosidase inhibitor and antidiabetic agent Miglitol at position C(2) creates an competitive inhibitor of five times higher activity than the parent compound. Its screening against a panel of human cell lines showed a low cytotoxicity therefore making this compound an interesting candidate for further clinical investigations.The synthesis of a monofluorinated α-glycosidase inhibitor from miglitol is reported.
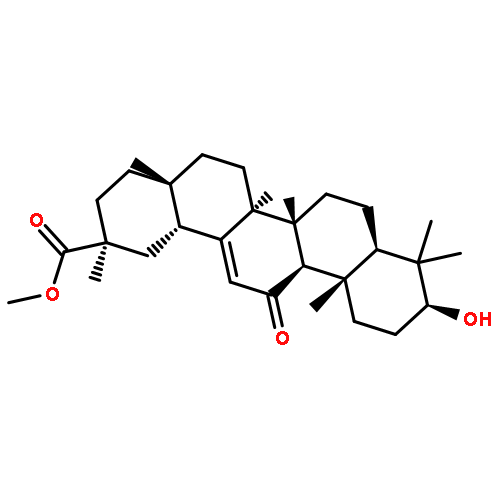
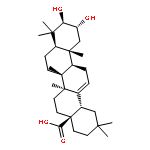
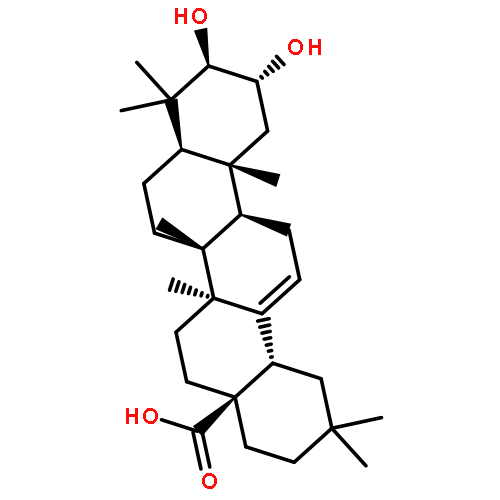
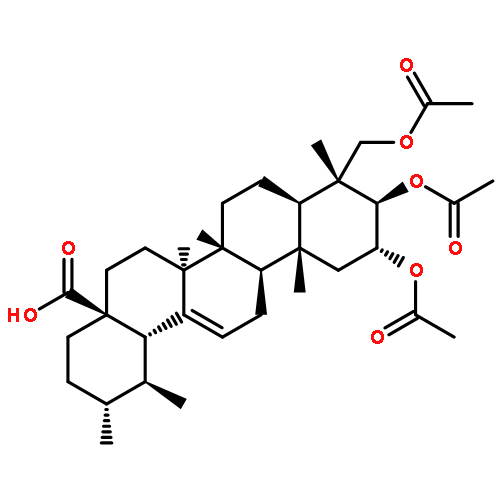
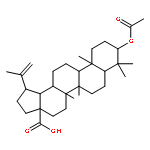
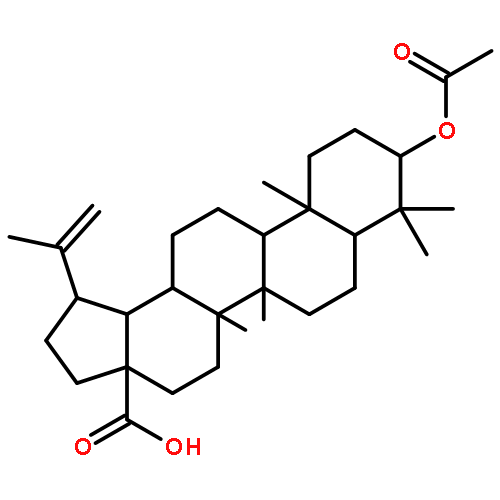
Co-reporter:Amalyn Nain-Perez, Luiz C.A. Barbosa, Diego Rodríguez-Hernández, Annemarie E. Kramell, Lucie Heller, René Csuk
Bioorganic & Medicinal Chemistry Letters (1 March 2017) Volume 27(Issue 5) pp:1141-1144
Publication Date(Web):1 March 2017
DOI:10.1016/j.bmcl.2017.01.079















![1,1'-[[(1alpha,2alpha,3beta,4beta)-2,4-bis(3,4,5-trimethoxyphenyl)cyclobutane-1,3-diyl]dicarbonyl]bis[5,6-dihydropyridin-2(1H)-one]](http://img.cochemist.com/ccimg/80300/80248-71-1.png)
![1,1'-[[(1alpha,2alpha,3beta,4beta)-2,4-bis(3,4,5-trimethoxyphenyl)cyclobutane-1,3-diyl]dicarbonyl]bis[5,6-dihydropyridin-2(1H)-one]](http://img.cochemist.com/ccimg/80300/80248-71-1_b.png)


![METHYL 1-AZABICYCLO[2.2.2]OCT-2-ENE-3-CARBOXYLATE](http://img.cochemist.com/ccimg/30500/30418-53-2.png)
![METHYL 1-AZABICYCLO[2.2.2]OCT-2-ENE-3-CARBOXYLATE](http://img.cochemist.com/ccimg/30500/30418-53-2_b.png)
![METHYL (1S,9R,10S,12S,13E)-13-ETHYLIDENE-10-HYDROXY-18-(HYDROXYMETHYL)-8,15-DIAZAPENTACYCLO[10.5.1.0<SUP>1,9</SUP>.0<SUP>2,7</SUP>.0<SUP>9,15</SUP>]OCTADECA-2,4,6-TRIENE-18-CARBOXYLATE](http://img.cochemist.com/ccimg/25500/25493-94-1.png)
![METHYL (1S,9R,10S,12S,13E)-13-ETHYLIDENE-10-HYDROXY-18-(HYDROXYMETHYL)-8,15-DIAZAPENTACYCLO[10.5.1.0<SUP>1,9</SUP>.0<SUP>2,7</SUP>.0<SUP>9,15</SUP>]OCTADECA-2,4,6-TRIENE-18-CARBOXYLATE](http://img.cochemist.com/ccimg/25500/25493-94-1_b.png)

















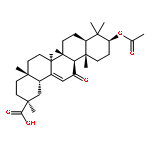










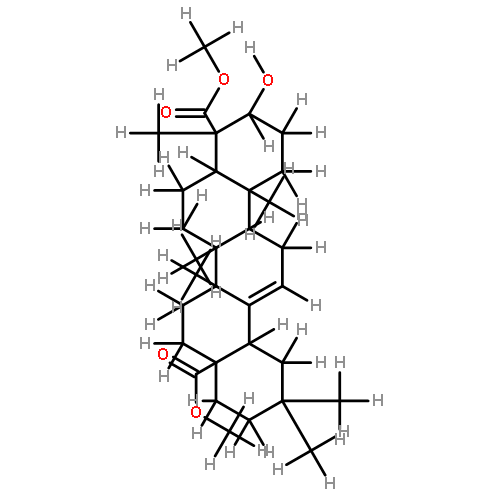


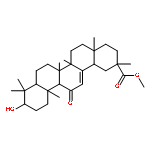









![[azido(phenyl)phosphoryl]benzene](http://img.cochemist.com/ccimg/4200/4129-17-3.png)
![[azido(phenyl)phosphoryl]benzene](http://img.cochemist.com/ccimg/4200/4129-17-3_b.png)




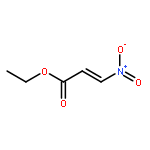









![[2,3'-Biindolinylidene]-2',3-dione](http://img.cochemist.com/ccimg/500/479-41-4.png)
![[2,3'-Biindolinylidene]-2',3-dione](http://img.cochemist.com/ccimg/500/479-41-4_b.png)