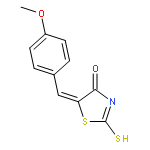
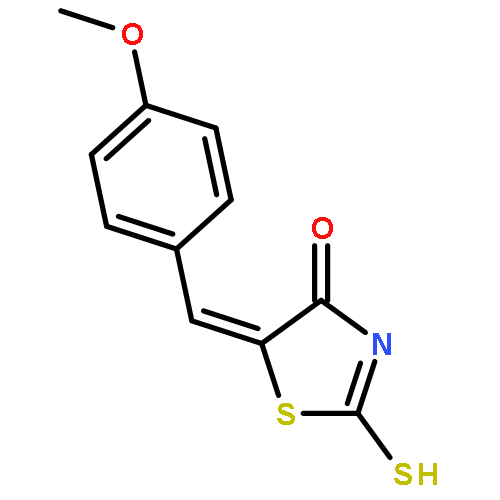
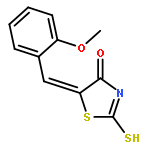
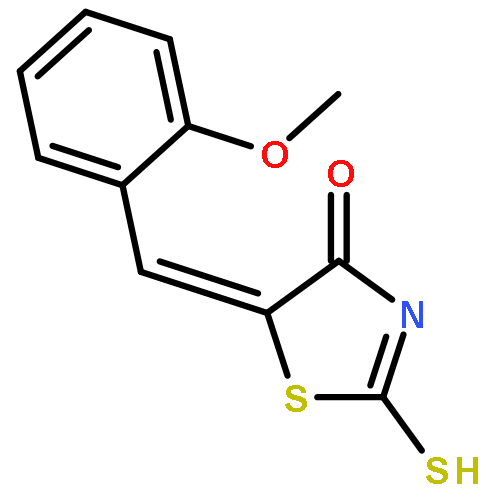
Co-reporter:Qinghua Hu, Mengze Zhou, Haoran Zhu, Guo Lu, Dongsen Zheng, Huanqiu Li, Kun Hao
Biomedicine & Pharmacotherapy 2017 Volume 86(Volume 86) pp:
Publication Date(Web):1 February 2017
DOI:10.1016/j.biopha.2016.12.002
Hyperuricemia is a kind of metabolic disease resulted from imbalance between urate production and excretion. Xanthine oxidase (XOD) or renal urate transporter 1 (URAT1) inhibitors have been applied for hyperuricemia treatment in clinic, but available drugs could not simultaneously target XOD and URAT1 and had various adverse effects. (E)-2-(4-bromophenyl)-1-(2, 4-dihydroxyphenyl)ethanone oxime (BDEO), as a deoxybenzoins oxime analog, was obtained from a cluster of deoxybenzoins derivatives synthesized by our research group with potent anti-hyperuricemic activity, which was expected to be dual inhibitor of XOD and URAT1. This study aimed to investigate effects of BDEO on XOD and URAT1 in vitro, as well as the possible mechanism by which BDEO attenuated hyperuricemia in vivo. In vitro, BDEO obviously inhibited XOD activity with an IC50 value of 3.33 μM, moreover, in Human embryonic kidney (HEK)293 cells expressing URAT1, BDEO and benzbromarone blocked uptake of uric acid with a Ki value of 0.145 μM. On the other hand, mice were orally administrated by oxonate for seven consecutive days to induce hyperuricemia, BDEO at various doses were administered intragastrically to hyperuricemic and normal mice daily. BDEO dose-dependently decreased serum urate level and uric acid excretion in 24 h in hyperuricemic mice. More importantly, BDEO significantly suppressed hepatic XOD activity and down-regulated renal URAT1 protein level in hyperuricemic mice. Notably, BDEO exhibited no effects on all these detected biochemical indicators in normal mice, predicting its safety. Taken together, the data suggested that BDEO may serve as a dual XOD and URAT1 inhibitor for treatment of hyperuricemia.
Co-reporter:Si-Ning Li, Yun-Yun Xu, Jia-Yu Gao, Hong-ran Yin, Shi-Lei Zhang, Huan-Qiu Li
Bioorganic & Medicinal Chemistry 2015 23(13) pp: 3221-3227
Publication Date(Web):
DOI:10.1016/j.bmc.2015.04.065
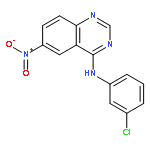
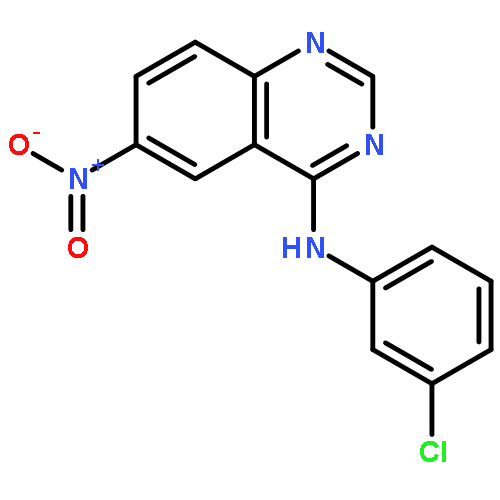
Co-reporter:Yun-Yun Xu, Si-Ning Li, Gao-Jian Yu, Qing-Hua Hu, Huan-Qiu Li
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 19) pp:6084-6091
Publication Date(Web):1 October 2013
DOI:10.1016/j.bmc.2013.06.070
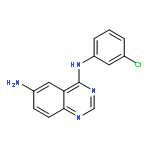
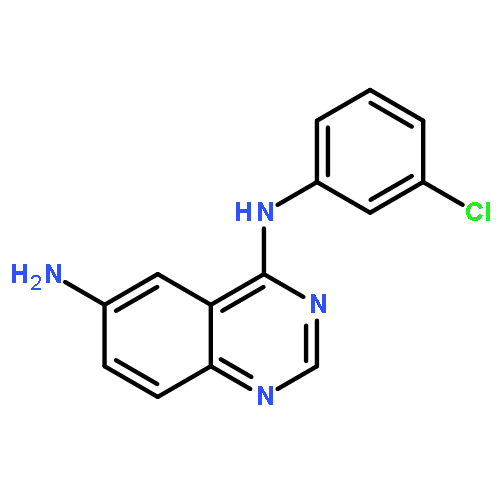
Two new series of new compounds containing a 6-amino-substituted group or 6-acrylamide-substituted group linked to a 4-anilinoquinazoline nucleus have been discovered as potential EGFR inhibitors. These compounds proved efficient effects on antiproliferative activity and EGFR–TK inhibitory activity. Especially, N6-((5-bromothiophen-2-yl)methyl)-N4-(3-chlorophenyl)quinazoline-4,6-diamine (5e), showed the most potent inhibitory activity (IC50 = 3.11 μM for Hep G2, IC50 = 0.82 μM for A549). The EGFR molecular docking model suggested that the new compound is nicely bound to the region of EGFR, and cell morphology by Hoechst stain experiment suggested that these compounds efficiently induced apoptosis of A549 cells.
Co-reporter:Yun-Yun Xu, Yi Cao, Hailkuo Ma, Huan-Qiu Li, Gui-Zhen Ao
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 2) pp:388-394
Publication Date(Web):15 January 2013
DOI:10.1016/j.bmc.2012.11.031
A type of novel α,β-unsaturated cyclohexanone analogous, which designed based on the curcumin core structure, have been discovered as potential EGFR inhibitors. These compounds exhibit potent antiproliferative activity in two human tumor cell lines (Hep G2 and B16-F10). Among them, compounds I3 and I12 displayed the most potent EGFR inhibitory activity (IC50 = 0.43 μM and 1.54 μM, respectively). Molecular docking of I12 into EGFR TK active site was also performed. This inhibitor nicely fitting the active site might well explain its excellent inhibitory activity.A type of novel α,β-unsaturated cyclohexanone analogous, which designed based on the curcumin core structure, have been discovered as potential EGFR inhibitors. These compounds exhibit potent antiproliferative activity in two human tumor cell lines (Hep G2 and B16-F10). Among them, compounds I3 and I12 displayed the most potent EGFR inhibitory activity (IC50 = 0.43 μM and 1.54 μM, respectively). Molecular docking of I12 into EGFR TK active site was also performed. This inhibitor nicely fitting the active site might well explain its excellent inhibitory activity.
Co-reporter:Huan-Qiu Li, Dong-Dong Li, Xiang Lu, Yun-Yun Xu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 1) pp:317-323
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmc.2011.10.085
A type of novel 4,6-substituted-(diaphenylamino)quinazolines, which designed based on the 4-(phenylamino)quinazoline moiety, have been discovered as potential EGFR inhibitors. These compounds displayed good antiproliferative activity and EGFR-TK inhibitory activity. Especially, 4-((4-(3-bromophenylamino)quinazolin-6-ylamino)methyl)phenol (5b), showed the most potent inhibitory activity (IC50 = 0.28 μM for Hep G2, IC50 = 0.59 μM for A16-F10 and IC50 = 0.87 μM for EGFR) and effectively induces apoptosis in a dose-dependent manner in the Hep G2 cell line. Molecular docking of 5b into EGFR TK active site was also performed. This inhibitor nicely fitting the active site might well explain its excellent inhibitory activity.A type of novel 4,6-substituted-(diaphenylamino)quinazolines, which designed based on the 4-(phenylamino)quinazoline moiety, have been discovered as potential EGFR inhibitors. These compounds displayed good antiproliferative activity and EGFR-TK inhibitory activity. Especially, 4-((4-(3-bromophenylamino)quinazolin-6-ylamino)methyl)phenol (5b), showed the most potent inhibitory activity (IC50 = 0.28 μM for Hep G2, IC50 = 0.59 μM for A16-F10 and IC50 = 0.87 μM for EGFR) and effectively induces apoptosis in a dose-dependent manner in the Hep G2 cell line. Molecular docking of 5b into EGFR TK active site was also performed. This inhibitor nicely fitting the active site might well explain its excellent inhibitory activity.
Co-reporter:Huan-Qiu Li, Yin Luo, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 15) pp:4454-4459
Publication Date(Web):1 August 2011
DOI:10.1016/j.bmc.2011.06.048
β-Ketoacyl-acyl carrier protein synthase III (FabH) catalyzes the initial step of fatty acid biosynthesis via a type II fatty acid synthase in most bacteria. The important role of this essential enzyme combined with its unique structural features and ubiquitous occurrence in bacteria has made it an attractive new target for the development of new FabH inhibitors. We first used a structure-based approach to develop 24 new vinylogous carbamates (4a–15a, 4b–15b) that target FabH for the development of new antibiotics in this paper. Potent FabH inhibitory and selective anti- Gram-negative bacteria activities were observed in most of these vinylogous carbamates. Especially, compound 6a and 7a showed the most potent FabH inhibitory activity with IC50 of 2.6 and 3.3 μM, respectively. Docking simulation was performed to position compound 6a into the Escherichia coli FabH active site and the possible binding conformation of compounds has been proposed. The biological data and molecular docking indicated that compounds 6a and 7a were potent inhibitors of E. coli FabH as antibiotics deserving further research.We first used a structure-based approach to develop 24 new vinylogous carbamates (4a–15a, 4b–15b) that target FabH for the development of new antibiotics in this paper. Compound 6a showed the most potent FabH inhibitory activity with IC50 of 2.6 μM, respectively. Docking simulation was performed to position compound 6a into the Escherichia coli FabH active site to determine the probable binding conformation.
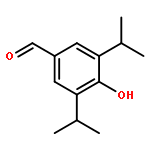
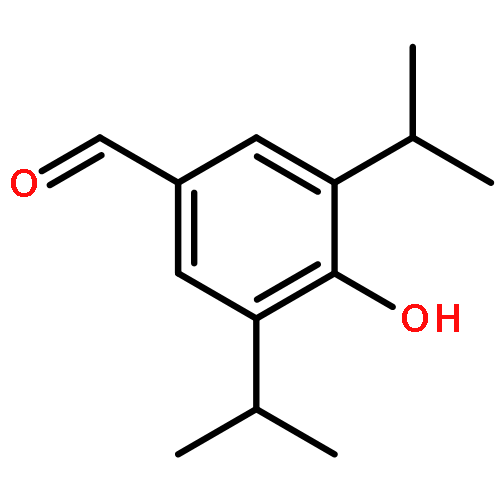
Co-reporter:Gui-Zhen Ao, Meng-Ze Zhou, Yu-Yao Li, Si-Ning Li, Hua-Nian Wang, Qian-Wen Wan, Huan-Qiu Li, Qing-Hua Hu
Bioorganic & Medicinal Chemistry (1 January 2017) Volume 25(Issue 1) pp:
Publication Date(Web):1 January 2017
DOI:10.1016/j.bmc.2016.10.022
A series of curcumin derivatives as potent dual inhibitors of xanthine oxidase (XOD) and urate transporter 1 (URAT1) was discovered as anti-hyperuricemic agents. These compounds proved efficient effects on anti-hyperuricemic activity and uricosuric activity in vivo. More importantly, some of them exhibited proved efficient effects on inhibiting XOD activity and suppressing uptake of uric acid via URAT1 in vitro. Especially, the treatment of 4d was demonstrated to improve uric acid over-production and under-excretion in oxonate-induced hyperuricemic mice through regulating XOD activity and URAT1 expression. Docking study was performed to elucidate the potent XOD inhibition of 4d. Compound 4d may serve as a tool compound for further design of anti-hyperuricemic drugs targeting both XOD and URAT1.