Co-reporter:Yuchen Zhou, Junjie Huang, Wei SunYuanlai Ju, Pinghui Yang, Lingyun Ding, Zhong-Ren Chen, Julia A. Kornfield
ACS Applied Materials & Interfaces 2017 Volume 9(Issue 4) pp:
Publication Date(Web):January 9, 2017
DOI:10.1021/acsami.6b13525
Patterned porous surfaces with responsive functionalities are fabricated by a thermoresponsive microgel-assisted breath figure (BF) process. When water droplets submerge into a polystyrene (PS) solution during formation of a porous surface by the bottom-up BF process, poly(N-isopropylacrylamide)-co-acrylic acid (PNIPAm-co-AA) microgels dispersed in the solution spontaneously assemble at the water–organic interfaces like “Pickering emulsions”, reinforced by capillary flow. The conformal layer of PNIPAm-co-AA microgels lining the pores appears in images from a scanning electron microscope (SEM) either as a smooth surface layer (L) or as an array of domelike protrusions (D), depending on the conditions at which the sample was dried for SEM. The change between L and D morphology correlates with the volume phase transition behavior of the microgels freely suspended: drying at a temperature below the volume phase transition temperature (VPTT) gives L, and the D morphology is formed by drying at a temperature greater than the VPTT of PNIPAm-co-AA microgels. The morphological transition is shown to accompany a significant change in surface contact angle (CA) relative to a corresponding pore layer made of PS, with L having a CA that is reduced by 85° relative to PS, while the decrease is only 22° for D. Porous structures with morphologically responsive surfaces could find application in biocatalysis or tissue engineering, for example, with functional enzymes sequestered when microgels are collaped and accessible when the microgels are swollen.Keywords: breath figure method; interfacial assembly of microgels; morphological transitions; responsive patterned polymer surfaces; wettability variations;
Co-reporter:Amy Fu, Kihak Gwon, Mihye Kim, Giyoong Tae, and Julia A. Kornfield
Biomacromolecules 2015 Volume 16(Issue 2) pp:
Publication Date(Web):December 23, 2014
DOI:10.1021/bm501543a
An in situ heparin-based forming hydrogel that cures under visible-light is formulated using eosin Y as a photoinitiator with triethanolamine as an electron donor to initiate reaction of thiolated-heparin with acrylate-ended poly(ethylene glycol). Formulations and irradiation conditions are presented for control of heparin content (1.6 to 3.3% w/v), modulus (100–10 000 Pa), and gelation time (30–600 s). Encapsulation of 3T3 fibroblasts in the hydrogel gave over 96% viability for all conditions examined. In vitro characterization of epidermal growth factor released from the hydrogel confirmed that the growth factor remains bioactive. The ability to deliver growth factors, fast gelation kinetics under visible light, and independent control of physical and biochemical properties makes this system a promising candidate for use in regenerative medicine. In particular, irradiation conditions that achieve gelation in 150s are compatible with the stringent light exposure limits of the retina, which affords a wide safety margin for use with other tissues.
Co-reporter:S. Ronca, G. Forte, A. Ailianou, J. A. Kornfield, and S. Rastogi
ACS Macro Letters 2012 Volume 1(Issue 9) pp:1116
Publication Date(Web):August 24, 2012
DOI:10.1021/mz300369x
The usual aggregation and precipitation driven by crystallization of nascent PE during homogeneous polymerization of ultra-high molecular weight polyethylene (UHMWPE) is inhibited by including linear low-density polyethylene (LLDPE) in the catalyst solution prior to addition of ethylene monomer. Co-crystallization of newly formed PE and dissolved LLDPE creates a polymer brush on the fold surfaces of the nascent crystallites. Consequently, aggregation is inhibited by steric stabilization. Scanning electron microscopy (SEM) images show that individual lamellae (approximately 10–20 nm thick) typically have lateral dimensions of 0.5 μm × 3.5 μm and form “bowtie” shaped stacks that are approximately 200–500 nm thick. This simple method for stabilizing nascent crystals against precipitation is enabling fundamental studies of their metastable “disentangled” state and may open scalable routes to compounding UHMWPE.
Co-reporter:Muzhou Wang
Journal of Biomedical Materials Research Part B: Applied Biomaterials 2012 Volume 100B( Issue 3) pp:618-623
Publication Date(Web):
DOI:10.1002/jbm.b.31981
Abstract
A method for evaluating strength of adhesives for hydrogels and soft tissues is presented. Quantitative measurements of shear strength for applications in tissue engineering and biomedicine are performed in torsion using a rheometer. Small, disk shaped specimens of soft biological tissues and/or hydrogels (8 mm diameter, 1–2 mm thick) are mounted onto rheometer tools and then bonded together using the adhesive to be tested. The torsional loading geometry imposes simple shear without deforming the planar adhesive bond, in contrast to the lap-shear test. It retains the advantages of the napkin ring test while reducing artifacts due to cutting and handling soft specimens. The method is demonstrated by measuring the shear strength of two types of biomedical adhesives (cyanoacrylate and polyethylene glycol-based) between model hydrogels (gelatin) and tissues (corneal stroma and skin). © 2012 Wiley Periodicals, Inc. J Biomed Mater Res Part B: Appl Biomater, 2012.
Co-reporter:Diana S. Smirnova, Julia A. Kornfield, and David J. Lohse
Macromolecules 2011 Volume 44(Issue 17) pp:6836-6848
Publication Date(Web):August 15, 2011
DOI:10.1021/ma200774u
Two-dimensional (2D) correlation analysis is applied to synchrotron X-ray scattering data to characterize morphological regimes during nonisothermal crystallization of a model ethylene copolymer (hydrogenated polybutadiene, HPBD). The 2D correlation patterns highlight relationships among multiple characteristics of structure evolution, particularly the extent to which separate features change simultaneously versus sequentially. By visualizing these relationships during cooling, evidence is obtained for two separate physical processes occurring in what is known as “irreversible crystallization” in random ethylene copolymers. Initial growth of primarily lamellae into unconstrained melt (“primary-irreversible crystallization”) is distinguished from subsequent secondary lamellae formation in the constrained, noncrystalline regions between the primary lamellae (“secondary-irreversible crystallization”). At successively lower temperatures (“reversible crystallization”), growth of the crystalline reflections is found to occur simultaneously with the change in shape of the amorphous halo, which is inconsistent with the formation of an additional phase. Rather, the synchronous character supports the view that growth of frustrated crystals distorts the adjacent noncrystalline material. Furthermore, heterocorrelation analysis of small-angle and wide-angle X-ray scattering data from the reversible crystallization regime reveals that the size of new crystals is consistent with fringed-micellar structures (∼9 nm). Thus, 2D correlation analysis provides new insights into morphology development in polymeric systems.
Co-reporter:Bradley D. Olsen, Julia A. Kornfield, and David A. Tirrell
Macromolecules 2010 Volume 43(Issue 21) pp:9094-9099
Publication Date(Web):October 13, 2010
DOI:10.1021/ma101434a
Injectable hydrogels show substantial promise for use in minimally invasive tissue engineering and drug delivery procedures. A new injectable hydrogel material, developed from recombinant telechelic proteins expressed in E. coli, demonstrates shear thinning by 3 orders of magnitude at large strains. Large-amplitude oscillatory shear illustrates that shear thinning is due to yielding within the bulk of the gel, and the rheological response and flow profiles are consistent with a shear-banding mechanism for yielding. The sharp yielding transition and large magnitude of the apparent shear thinning allow gels to be injected through narrow gauge needles with only gentle hand pressure. After injection the gels reset to full elastic strength in seconds due to rapid re-formation of the physical network junctions, allowing self-supporting structures to be formed. The shear thinning and recovery behavior is largely independent of the midblock length, enabling genetic engineering to be used to control the equilibrium modulus of the gel without loss of the characteristic yielding behavior. The shear-banding mechanism localizes deformation during flow into narrow regions of the gels, allowing more than 95% of seeded cells to survive the injection process.
Co-reporter:Yan Xia ; Bradley D. Olsen ; Julia A. Kornfield ;Robert H. Grubbs
Journal of the American Chemical Society 2009 Volume 131(Issue 51) pp:18525-18532
Publication Date(Web):November 30, 2009
DOI:10.1021/ja908379q
Efficient, one-pot preparation of synthetically challenging, high molecular weight (MW), narrowly dispersed brush block copolymers and random copolymers in high conversions was achieved by ring-opening metathesis (co)polymerization (ROMP) of various macromonomers (MMs) using the highly active, fast-initiating ruthenium olefin metathesis catalyst (H2IMes)(pyr)2(Cl)2RuCHPh. A series of random and block copolymers were prepared from a pair of MMs containing polylactide (PLA) and poly(n-butyl acrylate) (PnBA) side chains at similar MWs. Their self-assembly in the melt state was studied by small-angle X-ray scattering (SAXS) and atomic force microscopy (AFM). In brush random copolymers containing approximately equal volume fractions of PLA and PnBA, the side chains segregate into lamellae with domain spacing of 14 nm as measured by SAXS, which was in good agreement with the lamellar thickness measured by AFM. The domain spacings and order−disorder transition temperatures of brush random copolymers were insensitive to the backbone length. In contrast, brush block copolymers containing approximately equal volume fractions of these MMs self-assembled into highly ordered lamellae with domain spacing over 100 nm. Their assemblies suggested that the brush block copolymer backbone adopted an extended conformation in the ordered state.
Co-reporter:R.L. Ameri David, Ming-Hsin Wei, Julia A. Kornfield
Polymer 2009 50(26) pp: 6323-6330
Publication Date(Web):
DOI:10.1016/j.polymer.2009.10.032
Co-reporter:Neal R. Scruggs, Rafael Verduzco, David Uhrig, Waliullah Khan, Soo-Young Park, Jyotsana Lal and Julia A. Kornfield
Macromolecules 2009 Volume 42(Issue 1) pp:299-307
Publication Date(Web):December 5, 2008
DOI:10.1021/ma801598y
Diblock copolymers having a random-coil polymer block (polystyrene, PS) connected to a side-group liquid crystal polymer (SGLCP) self-assemble in a nematic liquid crystal (LC), 4-pentyl-4′-cyanobiphenyl, into micelles with PS-rich cores and SGLCP-rich coronas. The morphologies of block copolymers with varying PS content are characterized as a function of temperature and concentration using small-angle neutron scattering, rheometry, and transmission electron microscopy. Unlike conventional solvents, the nematic LC can undergo a first-order transition between distinct fluid phases, accessing the regimes of both strong and slight selectivity in a single polymer/solvent pair. Micelles dissolve away above a microphase separation temperature (MST) that is often equal to the solution’s isotropization point, TNI. However, increasing or decreasing the polymer’s PS content can shift the MST to be above or below TNI, respectively, and in the former case, micelles abruptly swell with solvent at TNI. Comparable effects can be achieved by modulating the overall polymer concentration.
Co-reporter:R. L. Ameri David, Ming-Hsin Wei, David Liu, Brett F. Bathel, Jan P. Plog, Albert Ratner and Julia A. Kornfield
Macromolecules 2009 Volume 42(Issue 4) pp:1380-1391
Publication Date(Web):February 3, 2009
DOI:10.1021/ma802058s
Solution properties are reported for homologous series of narrowly distributed polymers with systematically varied content of self-associating groups. Anionically polymerized polybutadienes of two lengths (510 and 1250 kg/mol) serve as prepolymers that are modified by incorporation of carboxylic acid side groups using thiol−ene coupling to pendant vinyl groups. Carboxylic acid groups strongly reduce polymer solubility in hydrocarbon solvents, restricting the extent of functionalization that can be examined in single-phase solutions (e.g., in chlorododecane, functionalization must be kept <1.8 mol % even for the shorter of the two backbones). In the single-phase regime, addition of hydrogen bond “stickers” weakly affects solution viscosity. Even at concentrations that produce overlap at the scale of strand length between stickers, viscosity increases are less than 1 order of magnitude. These controlled studies (using functionalized and unmodified polymer homologues of matched, well-defined length) challenge the pre-existing understanding of the rheology of self-associating polymers. The results indicate that effects of intrachain pairing are important beyond the dilute regime—behavior unaccounted for in earlier experimental and theoretical studies. The implications for mist control of aviation fuel are that self-associating polymers of acceptable solubility in the fuel are not superior to nonassociating polymers even at concentrations several times above overlap.
Co-reporter:Wei Shen, Julia A. Kornfield and David A. Tirrell
Soft Matter 2007 vol. 3(Issue 1) pp:99-107
Publication Date(Web):09 Nov 2006
DOI:10.1039/B610986A
Artificial protein hydrogels made from a triblock protein (designated AC10A, where A is an acidic zipper domain and C10 comprises 10 repeats of the nonapeptide sequence exhibit normalized plateau storage moduli (G′∞/nkT) less than 0.13 at all concentrations, pH values, and ionic strengths examined. These gels are surprisingly soft due to loop formation at the expense of bridges between physical junctions. Molecular-level evidence of loop formation is provided by strong fluorescence energy transfer (FRET) between distinct chromophores placed at the C- and N-termini of labelled chains diluted in an excess of unlabelled chains. The tendency to form loops originates from the compact size of the random coil midblock (mean RH(C10) ≈ 20 Å, determined from quasi-elastic light scattering of C10), and is facilitated by the ability of the leucine zipper domains to form antiparallel aggregates. Although the aggregation number of the leucine zipper domains is small (tetrameric, determined from multi-angle static light scattering of AC10 diblock), the average center-to-center distance between aggregates is roughly 1.5 times the average end-to-end distance of the C10 domain in a 7% w/v network. To avoid stretching the C10 domain, the chains tend to form loops. Changes in pH or ionic strength that expand the polyelectrolyte midblock favor bridging, leading to greater G′∞ as long as leucine zipper endblocks do not dissociate. Understanding of the network structure provided successful design strategies to increase the rigidity of these hydrogels. In contrast to intuitive design concepts for rubber and gel materials, it was shown that increasing either the length or the charge density of the midblock increases rigidity, because fewer chains are wasted in loop formation.
Co-reporter:Rafael Verduzco, Neal R. Scruggs, Samuel Sprunt, Peter Palffy-Muhoray and Julia A. Kornfield
Soft Matter 2007 vol. 3(Issue 8) pp:993-1002
Publication Date(Web):30 May 2007
DOI:10.1039/B700871F
Nematic liquid-crystal (LC) elastomers and gels have a rubbery polymer network coupled to the nematic director. While LC elastomers show a single, non-hydrodynamic relaxation mode, dynamic light-scattering studies of self-assembled liquid-crystal gels reveal orientational fluctuations that relax over a broad time scale. At short times, the relaxation dynamics exhibit hydrodynamic behavior. In contrast, the relaxation dynamics at long times are non-hydrodynamic, highly anisotropic, and increase in amplitude at small scattering angles. We argue that the slower dynamics arise from coupling between the director and the physically associated network, which prevents director orientational fluctuations from decaying completely at short times. At long enough times the network restructures, allowing the orientational fluctuations to fully decay. Director dynamics in the self-assembled gels are thus quite distinct from those observed in LC elastomers in two respects: they display soft orientational fluctuations at short times, and they exhibit at least two qualitatively distinct relaxation processes.
Co-reporter:Julia A. Kornfield;Neal R. Scruggs
Macromolecular Chemistry and Physics 2007 Volume 208(Issue 19‐20) pp:2242-2253
Publication Date(Web):5 OCT 2007
DOI:10.1002/macp.200700342
Addition of a small-molecule liquid crystal (5CB) to a cyanobiphenyl-based side-group liquid crystal polymer (SGLCP) stabilizes nematic order, increasing the isotropization temperature (TNI) more than 15 °C. Despite synergistic ordering at high concentration, small amounts of polymer destabilize nematic order. Even though TNI(SGLCP) is 27 °C greater than TNI(5CB), 2H NMR shows that the order parameter of the SGLCP is less than that of 5CB at concentrations for which monodomains were accessible (≤10 wt.-%). The results imply that nematic order is frustrated in the bulk polymer and addition of small molecule LC relaxes this frustration by allowing greater configurational freedom. Conversely, adding small amounts of polymer to the bulk 5CB introduces frustration, resulting in the strong asymmetry of the phase diagram.
Co-reporter:Michael D. Kempe, Rafael Verduzco, Neal R. Scruggs and Julia A. Kornfield
Soft Matter 2006 vol. 2(Issue 5) pp:422-431
Publication Date(Web):04 Apr 2006
DOI:10.1039/B600483K
Rheological properties of triblock copolymers dissolved in a nematic liquid crystal (LC) solvent demonstrate that their microphase separated structure is heavily influenced by changes in LC order. Nematic gels were created by swelling a well-defined, high molecular weight ABA block copolymer with the small-molecule nematic LC solvent 4-pentyl-4′-cyanobiphenyl (5CB). The “B” midblock is a side-group liquid crystal polymer (SGLCP) designed to be soluble in 5CB and the “A” endblocks are polystyrene, which is LC-phobic and microphase separates to produce a physically cross-linked, thermoreversible, macroscopic polymer network. At sufficiently low polymer concentration a plateau modulus in the nematic phase, characteristic of a gel, abruptly transitions to terminal behavior when the gel is heated into its isotropic phase. In more concentrated gels, endblock aggregates persist into the isotopic phase. Dramatic changes in network structure are observed over small temperature windows (as little as 1 °C) due to tccche rapidly changing LC order near the isotropization point. The discontinuous change in solvent quality produces an abrupt change in viscoelastic properties for three polymers having different pendant mesogenic groups and matched block lengths.
Co-reporter:Charles S. Nickerson;Hampar L. Karageozian;John Park
Macromolecular Symposia 2005 Volume 227(Issue 1) pp:183-190
Publication Date(Web):4 AUG 2005
DOI:10.1002/masy.200550918
The vitreous humor is a soft viscoelastic gel composed of a complex network of biomolecules (primarily collagen and polyhyaluronic acid, “hyaluronan”). While extensive progress has been made in identifying the components of the vitreous, lack of sufficient experimental methods has hampered previous efforts to quantitatively define its mechanical properties. To address this issue, we have developed a novel “cleat” tool geometry for dynamic shear rheometry. Using this geometry we find that the shear moduli of vitreous decline by roughly a factor of five to steady-state values within an hour after removal from the eye. Steady-state values (Porcine: G′ = 2.6 ± 0.9 Pa, and G″ = 0.65 ± 0.41 Pa, n = 9; Bovine: G′ = 6.5 ± 2.0 Pa, and G″ = 2.0 ± 0.6 Pa, n = 17) are significantly larger than previously reported. The decrease in modulus also correlates with a decrease in mass (65 ± 11 %, n = 8) that occurs spontaneously after the vitreous is extracted. Rheological findings, taken in combination with biochemical analyses suggest that hyaluronan, a ubiquitous polyelectrolyte in connective tissue, contributes to vitreous stiffness by inducing tension in the network in vivo.
Co-reporter:Giyoong Tae, Julia A. Kornfield, Jeffrey A. Hubbell
Biomaterials 2005 Volume 26(Issue 25) pp:5259-5266
Publication Date(Web):September 2005
DOI:10.1016/j.biomaterials.2005.01.042
Poly(ethylene glycol)s modified with fluorocarbon end groups are capable of in situ transition from an injectable liquid to a viscoelastic hydrogel by hydrophobic interaction of the end groups; this class of materials is useful for a variety of biomedical applications, including sustained protein release. The hydrogel state can be transformed into an injectable state by the addition of a toxicologically acceptable organic solvent, such as N-methyl pyrrolidone; after injection, this solution quickly returns to a gel state by diffusion of the water-miscible organic solvent into the surrounding environment. In vitro characterization of sustained release of human growth hormone (hGH) using this injectable depot shows that hGH remains stable inside the hydrogel formed, and demonstrates more than 2 weeks of prolonged release of hGH complexed with Zn2+ ions without protein aggregation or initial burst.
Co-reporter:G. Tae;R.G.H. Lammertink;J.A. Kornfield;J.A. Hubbell
Advanced Materials 2003 Volume 15(Issue 1) pp:
Publication Date(Web):10 JAN 2003
DOI:10.1002/adma.200390013
Co-reporter:Charles S. Nickerson, John Park, Julia A. Kornfield, Hampar Karageozian
Journal of Biomechanics (2008) Volume 41(Issue 9) pp:1840-1846
Publication Date(Web):1 January 2008
DOI:10.1016/j.jbiomech.2008.04.015
Coordinated rheological and biochemical measurements provide the linear and nonlinear mechanical properties of the vitreous and demonstrate the structural role of hyaluronic acid. “Cleated” tools are used to overcome wall slip and avoid tissue compression during measurements of the dynamic moduli of fresh porcine and bovine vitreous. Shear moduli decreased five-fold from initial to steady-state values in the first hour after dissection. Steady-state values (porcine: G′=2.8±0.9 Pa, n=9; bovine: G′=7.0±2.0 Pa, n=17) are significantly greater than previously reported. The decrease in modulus after removal from the eye correlates with a decrease in mass: even in the absence of external driving forces, porcine vitreous expels ∼5% of its mass within 5 min and continues to decay to a steady-state mass ∼10% lower than its initial mass. The expelled fluid has a substantial hyaluronan concentration, but very low protein content. These results indicate that the vitreous network is under tension at its native volume and its high initial modulus results from this state of tension. We hypothesize that hyaluronan plays a role in sustaining the “internal tension” by Donnan swelling.
Co-reporter:Miao Hu ; Yan Xia ; Gregory B. McKenna ; Julia A. Kornfield ;Robert H. Grubbs
Macromolecules () pp:
Publication Date(Web):August 4, 2011
DOI:10.1021/ma2009673
We have examined the linear rheological responses of a series of well-defined, dense, regularly branched brush polymers. These narrow molecular weight distribution brush polymers had polynorobornene backbones with degrees of polymerization (DP) of 200, 400, and 800 and polylactide side chains with molecular weight of 1.4 kDa, 4.4 kDa, and 8.7 kDa. The master curves for these brush polymers were obtained by time temperature superposition (TTS) of the dynamic moduli over the range from the glassy region to the terminal flow region. Similar to other long chain branched polymers, these densely branched brush polymers show a sequence of relaxation. Subsequent to the glassy relaxation, two different relaxation processes can be observed for samples with the high molecular weight (4.4 and 8.7 kDa) side chains, corresponding to the relaxation of the side chains and the brush polymer backbone. Influenced by the large volume fraction of high molecular weight side chains, these brush polymers are unentangled. The lowest plateau observed in the dynamic response is not the rubbery entanglement plateau but is instead associated with the steady state recoverable compliance. Side chain properties affect the rheological responses of these densely branched brush polymers and determine their glassy behaviors.
.jpg)
![1,2,3,4,5-CYCLOPENTANEPENTAYL, COMPD. WITH 1-[(1S)-1-(DIBICYCLO[2.2.1]HEPT-2-YLPHOSPHINO)ETHYL]-2-[2-(DIPHENYLPHOSPHINO)PHENYL]-1,2,3,4,5-CYCLOPENTANEPENTAYL, IRON SALT (1:1:1)](http://img.cochemist.com/ccimg/134000/133978-15-1.png)
![1,2,3,4,5-CYCLOPENTANEPENTAYL, COMPD. WITH 1-[(1S)-1-(DIBICYCLO[2.2.1]HEPT-2-YLPHOSPHINO)ETHYL]-2-[2-(DIPHENYLPHOSPHINO)PHENYL]-1,2,3,4,5-CYCLOPENTANEPENTAYL, IRON SALT (1:1:1)](http://img.cochemist.com/ccimg/134000/133978-15-1_b.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2_b.png)






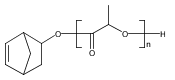







![Pyrimidine, 2-[4-(9-decenyloxy)phenyl]-5-decyl-](http://img.cochemist.com/ccimg/117600/117584-39-1.png)
![Pyrimidine, 2-[4-(9-decenyloxy)phenyl]-5-decyl-](http://img.cochemist.com/ccimg/117600/117584-39-1_b.png)
![Pyrimidine, 5-decyl-2-[4-(7-octenyloxy)phenyl]-](http://img.cochemist.com/ccimg/117600/117560-68-6.png)
![Pyrimidine, 5-decyl-2-[4-(7-octenyloxy)phenyl]-](http://img.cochemist.com/ccimg/117600/117560-68-6_b.png)