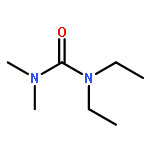
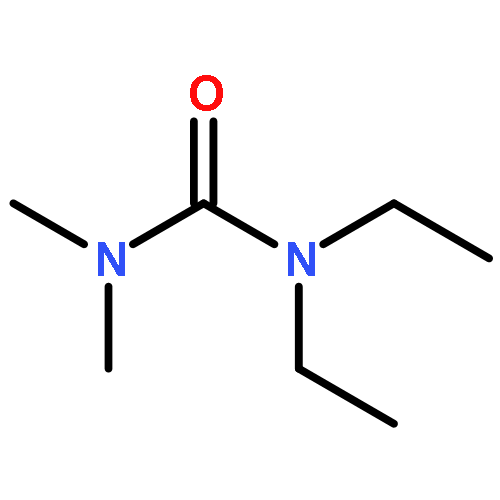
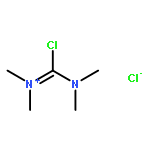
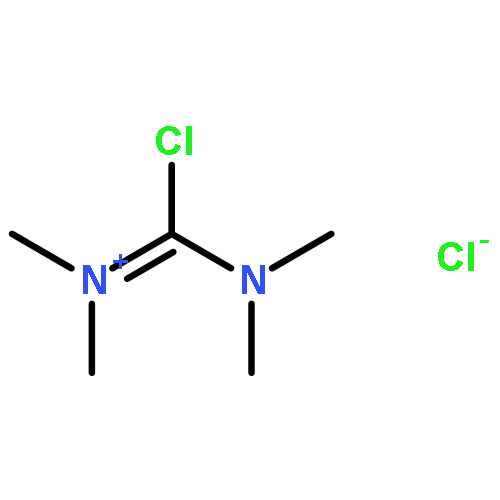
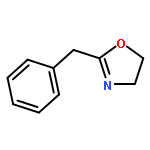
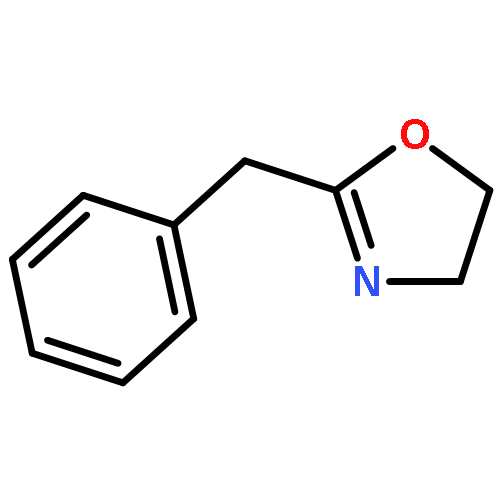
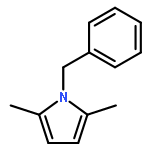
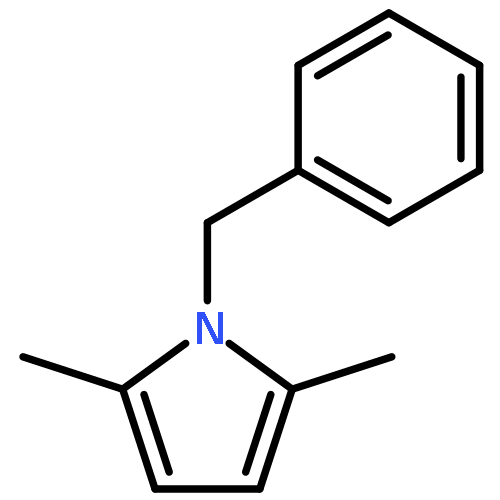
Co-reporter:Philipp Kratzer, Bastian Ramming, Steven Römisch and Gerhard Maas
CrystEngComm 2015 vol. 17(Issue 24) pp:4486-4494
Publication Date(Web):15 May 2015
DOI:10.1039/C5CE00365B
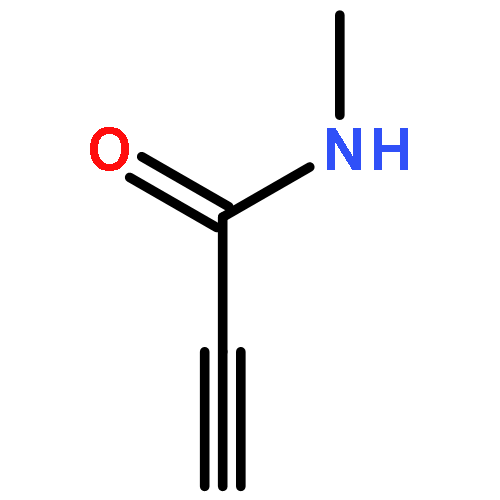
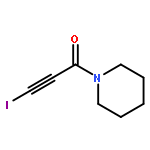
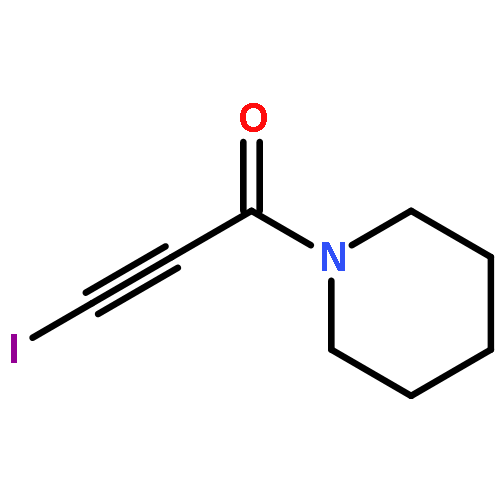
The solid-state structures of six 3-iodopropiolamides (I–CC–CO–NR2; NR2 = pyrrolidino, 2,5-dihydro-1H-pyrrolo, piperidino, morpholino, methylamino, amino) have been determined by XRD analysis. All but the parent 3-iodopropiolamide exhibit very short intermolecular iodine–oxygen contacts (2.754(2)–2.858(5) Å) leading to one-dimensional polymeric chains in the solid state. The shape of these chains and the packing arrangement depend on the size and ring conformation in the cases of cyclic tertiary amides (NR2). In the structure of 3-iodopropiol-N-methylamide, I⋯O halogen bonds and N–H⋯O hydrogen bonds generate a two-dimensional, non-planar infinite network, while the crystal structure of 3-iodopropiolamide acetonitrile solvate is determined by a network of hydrogen bonds involving the CONH2 moiety; the iodine atom, on the other hand, is coordinated to an acetonitrile molecule through a short I⋯N contact.
Co-reporter:Maria Arkhipova, Svetlana Eichel and Gerhard Maas
RSC Advances 2014 vol. 4(Issue 99) pp:56506-56517
Publication Date(Web):27 Oct 2014
DOI:10.1039/C4RA09567G
The solubility of titanium and aluminium alcoholates and of titanium tetrakis(trimethylsilanolate) in several hexaalkylguanidinium-based room temperature ionic liquids was screened. The solvent/solute combinations which displayed the highest alcoholate solubility and stability were applied as Lewis-acidic catalytic media for several dehydrating cyclocondensations: lactamisation of ω-aminocarboxylic acids, direct amidation of carboxylic acids, synthesis of oxazolines from carboxylic acids and 2-aminoethanol, lactonisation of 6-hydroxyhexanoic acid, and Paal–Knorr synthesis of pyrroles.
Co-reporter:Werner Weingärtner
European Journal of Organic Chemistry 2012 Volume 2012( Issue 32) pp:6372-6382
Publication Date(Web):
DOI:10.1002/ejoc.201201005
Abstract
Thermal and microwave-assisted [3+2] cycloadditions between differently substituted propiolamidinium tetraphenylborates 3a–d and N-methyl-C-phenylnitrone, benzyl azide, and N-(3-azidopropyl)phthalimide were studied. The activation parameters of the [3+2] cycloaddition between alkyne 3a and benzyl azide were determined. A Diels–Alder reaction between the terminal alkyne 3a and cyclopentadiene could be achieved with the aid of microwave activation. The reaction between 3a and triphenylphosphorane imine provides the β,β-bis(dimethylamino)vinylphosphonium salt 21, which might or might not have been formed through an initial [2+2] cycloaddition reaction.
Co-reporter:Ahmed El-Gokha, Gerhard Maas
Tetrahedron 2011 67(16) pp: 2849-2857
Publication Date(Web):
DOI:10.1016/j.tet.2011.02.068
Co-reporter:Stefan Maurer;Christian Burkhart
European Journal of Organic Chemistry 2010 Volume 2010( Issue 13) pp:2504-2511
Publication Date(Web):
DOI:10.1002/ejoc.201000102
Abstract
Phosphirano[1,2-c][1,2,3]diazaphospholes 5, which feature a strongly pyramidalized phosphorus atom at a ring fusion position, were identified as a new type of phosphane ligands for transition metals. The derivatives 5a–c smoothly form W(CO)5 complexes, which are thermally quite stable – in contrast to the known lability of various carbonylmetal complexes of simple phosphiranes. In the temperature range 120–150 °C, they undergo a clean decomplexation in toluene solution. Bicyclic phosphiranes 5a and 5b, but not the acceptor-substituted derivative 5c, readily react with the [(E,E)-dibenzylideneacetone]palladium(0) complex Pd2(dba)3·CHCl3 to form the corresponding isolable complexes [(5a)2(dba)Pd] and [(5b)2(dba)Pd]. In agreement with the formation of these complexes, it was found that 5a,b as ligands effectively promote the Suzuki cross-coupling of 4-bromotoluene with benzeneboronic acid. The solid-state structure of bicyclic phosphirane 5b and of the complexes [(5a)(CO)5W], [(5b)2(dba)Pd] (pentane solvate) and [(5b)2(dba)Pd] (benzene solvate) were determined by X-ray diffraction analysis.
Co-reporter:Stefan Maurer;Tamaki Jikyo
European Journal of Organic Chemistry 2009 Volume 2009( Issue 13) pp:2195-2207
Publication Date(Web):
DOI:10.1002/ejoc.200900002
Abstract
Spiro[fluorene-9,6′-[2]thia[1]phosphabicyclo[3.1.0]hex[3]enes] 7a–c have been obtained in one step from 3,5-diaryl-1,2-thiaphospholes and 9-diazofluorene or its 2,7-dibromo derivative. The bicyclic phosphiranes are stable against water and resist attempts at sulfuration or selenation of the phosphorus atom. However, cleavage of the P–C ring fusion with hydrogen chloride followed by hydrolysis led to the monocyclic 2-(9H-fluoren-9-yl)-2,3-dihydro-1,2-thiaphosphole 2-oxides 8a–c. Phosphiranes 7a–c also react to form the hexacarbonyltungsten-(P–W) complexes 10a–c readily and in high yields. These complexes rearrange in toluene solution at 50–80 °C to form the spiro[fluorene-9,2′-[1]phospha[6]thiabicyclo[3.1.0]hex-3-ene]-W(CO)5-(P–W) complexes 11a–c, which are the first representatives of ring-fused thiaphosphiranes. Compound 11a is readily desulfurated with tributylphosphane to form the highly oxygen-sensitive spiro[fluorene-9,2′-[2H]phosphole] 13, which is reconverted into 11a upon treatment with sulfur. Bicyclic 1,3,2-dithiaphospholanes 12a–c are formed as minor byproducts of the thermal isomerization of phosphiranes 10. Compound 10a slowly rearranges in solution to give 12a as the sole product. Compounds 12 may result from a formal [3+2] cycloaddition reaction of an α,β-unsaturated thione, formed by partial decomposition of 10, with a dipolar valence isomer of 10 or 11, featuring aW(CO)5-complexed thiocarbonyl-phospha-ylide dipole (R2C=S+–P–R). W(CO)5 complexation is not a prerequisite for the cycloaddition reaction: the uncomplexed phosphirano-thiaphosphole 7a reacts with thiobenzophenone in the same way to form the bicyclic 1,3,2-dithiaphospholane 18 in high yield. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Gerhard Maas Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 44) pp:8186-8195
Publication Date(Web):
DOI:10.1002/anie.200902785
Abstract
Diazo compounds (R1R2CN2) are known as versatile and useful substrates for an array of chemical transformations and, therefore, diazo chemistry is still far from losing anything of its long-standing fascination. In addition to many studies on the subsequent chemistry of the diazo group, the inventory of methods for the preparation of diazo compounds is continuously supplemented by new methods and novel variations of established procedures. Several of these synthetic approaches take into account the lability and remarkable chemical reactivity of certain classes of diazo compounds, and environmentally more benign procedures also continue to be developed.
Co-reporter:Gerhard Maas, Andreas Müller, Andreas Endres, Jürgen Schatz
Tetrahedron 2009 65(29–30) pp: 5733-5738
Publication Date(Web):
DOI:10.1016/j.tet.2009.05.014
Co-reporter:Gerhard Maas Dr.
Angewandte Chemie 2009 Volume 121( Issue 44) pp:8332-8341
Publication Date(Web):
DOI:10.1002/ange.200902785
Abstract
Aufgrund der vielseitigen und nützlichen Folgechemie von Diazoverbindungen (R1R2CN2) ist das Interesse an dieser Substanzklasse seit Jahren ungebrochen. Dabei wird auch das bereits vorhandene Methodenrepertoire zur Herstellung von Diazoverbindungen immer wieder um neue Verfahren oder um neuartige Varianten etablierter Synthesewege ergänzt. Nicht selten tragen die neuen Synthesevorschläge der Labilität und beachtlichen chemischen Reaktivität bestimmter Typen von Diazoverbindungen Rechnung, aber auch dem Aspekt umweltfreundlicherer Methodik wurde Beachtung geschenkt.
Co-reporter:Stefan M. Bucher;Ralf Brückmann
European Journal of Organic Chemistry 2008 Volume 2008( Issue 26) pp:4426-4433
Publication Date(Web):
DOI:10.1002/ejoc.200800524
Abstract
Aryl-substituted α-silyl α-diazo ketones are readily transformed into aryl silyl ketenes in the presence of a catalytic amount of triflic acid. Thus, a convenient method to prepare these silyl ketenes becomes available, which combines two steps, silylation of an aryl diazomethyl ketone and acid-induced Wolff rearrangement of the formed α-silyl α-diazo ketone, in a one-pot procedure. It appears that the trialkylammonium salt, which is formed in the silylation step, can also catalyze the Wolff rearrangement, but distinctly more slowly than the proton acid. The silyl ketenes react smoothly with α-silyl α-diazo ketones to form 3-silyl-1-silyloxyallenes in fairly good yields. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Lutz Schäffler, Stefan Buck, Gerhard Maas
Inorganica Chimica Acta 2008 Volume 361(Issue 1) pp:109-122
Publication Date(Web):1 January 2008
DOI:10.1016/j.ica.2007.06.016
The dinuclear bis(6-X-pyridin-2-olato) ruthenium complexes [Ru2(μ-XpyO)2(CO)4(PPh3)2] (X = Cl (4B) and Br (5B)), [Ru2(μ-XpyO)2(CO)4(CH3CN)2] (X = Cl (6B), Br (7B) and F (8B)) and [Ru2(μ-ClpyO)2(CO)4(PhCN)2] (9B) were prepared from the corresponding tetranuclear coordination dimers [Ru2(μ-XpyO)2(CO)4]2 (1: X = Cl; 2: X = Br) and [Ru2(μ-FpyO)2(CO)6]2 (3) by treatment with an excess of triphenylphosphane, acetonitrile and benzonitrile, respectively. In the solid state, complexes 4B–9B all have a head-to-tail arrangement of the two pyridonate ligands, as evidenced by X-ray crystal structure analyses of 4B, 6B and 9B, in contrast to the head-to-head arrangement in the precursors 1–3. A temperature- and solvent-dependent equilibrium between the yellow head-to-tail complexes and the red head-to-head complexes 4A–7A and 9A, bearing an axial ligand only at the O,O-substituted ruthenium atom, exists in solution and was studied by NMR spectroscopy. Full 1H and 13C NMR assignments were made in each case. Treatment of 1 and 2 with the N-heterocyclic carbene (NHC) 1-butyl-3-methylimidazolin-2-ylidene provided the complexes [Ru2(μ-XpyO)2(CO)4(NHC)], X = Cl (11A) or Br (12A). An XRD analysis revealed the head-to-head arrangement of the pyridonate ligands and axial coordination of the carbene ligand at the O,O-substituted ruthenium atom. The conversion of 11A and 12A into the corresponding head-to-tail complexes was not possible.The diruthenium(I,I) complexes shown in the Scheme have a head-to-tail arrangement of the two pyridonate ligands in the solid state. In solution, a temperature- and solvent-dependent equilibrium between head-to-tail and head-to-head complexes exists which was studied by NMR spectroscopy. When L is an imidazolylidene ligand, a stable head-to-head arrangement is found where only the O,O-substituted Ru atom bears a carbene ligand.
Co-reporter:Markus Grohmann;Stefan Buck;Lutz Schäffler
Advanced Synthesis & Catalysis 2006 Volume 348(Issue 15) pp:
Publication Date(Web):12 OCT 2006
DOI:10.1002/adsc.200606108
Intramolecular carbenoid CH insertion of five α-diazoacetamides [N2CHCONR2, NR2=NEt2 (3a), NBu2 (3b), N(i-Pr)2 (3c), N(CH2Ph)2 (3d), N(i-Pr)(CH2Ph) (3e)], was investigated using as catalysts dinuclear Ru(I,I) complexes of the type [Ru2(μ-L1)2(CO)4L22], where L1 is a bidentate bridging acetate, calix[4]arenedicarboxylate, saccharinate, pyridin-2-olate, or triazenide ligand, as well as [RuCl2(p-cymene)]2. The Ru(I,I) complexes were found to be suitable catalysts for the carbenoid cyclization reactions, except in the case of 3a. With diazoamides 3b–e, [Ru2(μ-sac)2(CO)5]2 (sac=saccharinate) and [Ru2(μ-6-chloropyridin-2-olate)2(CH3CN)2(CO)4] are as effective as Rh2(OAc)4 under the same conditions, although some differences in the regioselectivity and chemoselectivity of the cyclization are observed. The carbenoid cyclization reactions yield γ-lactams from diazoamides 3a and 3b, both a β- and a γ-lactam from 3c, and a β-lactam as well as a 3-azabicyclo[5.3.0]deca-5,7,9-trien-2-one from 3d. With 3e, formation of γ-lactam 21 and of bicyclic lactam 23 prevails.
Co-reporter:Lutz Schäffler, Bernhard Müller, Gerhard Maas
Inorganica Chimica Acta 2006 Volume 359(Issue 3) pp:970-977
Publication Date(Web):1 February 2006
DOI:10.1016/j.ica.2005.07.023
The complexes [Ru2(CO)5(μ-FpyO)2]2 (1), [Ru2(CO)4(μ-ClpyO)2]2 (2), and [Ru2(CO)4(μ-BrpyO)2]2 (3) were prepared from Ru3(CO)12 and 6-fluoro-2-hydroxypyridine (FpyOH), 6-chloro-2-hydroxypyridine (ClpyOH) and 6-bromo-2-hydroxypyridine (BrpyOH), respectively, in hot toluene. Compounds 1–3 are coordination dimers with a cyclo-RuORuO motif. By carrying out the reaction in hot methanol, the dinuclear complexes [Ru2(CO)4(μ-ClpyO)2(CH3OH)] (4) and [Ru2(CO)4(μ-BrpyO)2(CH3OH)] (5), respectively, were obtained. Treatment of 2 and 3 with triphenylphosphane provided the complexes [Ru2(CO)4(μ-ClpyO)2(PPh3)] (6) and [Ru2(CO)4(μ-BrpyO)2(PPh3)] (7), respectively. The solid-state structures of complexes 1, 2, 4, 6, and 7 were determined by single crystal X-ray diffraction. In all cases, a head–head coordination of the two 6-halopyridinolate ligands at the Ru22+ core was found. In all chlorine- or bromine-containing complexes, the axial coordination site at the ruthenium atom neighbored by two Cl or Br atoms remains unoccupied due to steric shielding by the halogen atom. In the fluoropyridinolate complex 1, the same coordination site is occupied by a carbonyl ligand.The preparation and solid-state structures of several ruthenium complexes of the types [Ru2(CO)5(μ-FpyO)2]2 and [Ru2(CO)4(μ-HalpyO)2(L)] (Hal = Cl, Br; L = CH3OH, PPh3) are described.
Co-reporter:Thorsten Werle, Lutz Schäffler, Gerhard Maas
Journal of Organometallic Chemistry 2005 Volume 690(24–25) pp:5562-5569
Publication Date(Web):1 December 2005
DOI:10.1016/j.jorganchem.2005.06.043
Dinuclear ruthenium(I,I) carboxylate complexes [Ru2(CO)4(μ-OOCR)2]n (R = CH3 (1a), C3H7 (1b), H (1c), CF3 (1d)) and 2-pyridonate complex [Ru2(CO)4(μ-2-pyridonate)2]n (3) catalyze efficiently the cyclopropanation of alkenes with methyl diazoacetate. High yields are obtained with terminal nucleophilic alkenes (styrene, ethyl vinyl ether, α-methylstyrene), medium yields with 1-hexene, cyclohexene, 4,5-dihydrofuran and 2-methyl-2-butene. The E-selectivity of the cyclopropanes obtained from the monosubstituted alkenes and the cycloalkenes decreases in the order 1b > 1a > 1d > 1c. The cyclopropanation of 2-methyl-2-butene is highly syn-selective. Several complexes of the type [Ru2(CO)4(μ-L1)2]2 (4) and (5), [Ru2(CO)4(μ-L1)2L2] (L2 = CH3OH, PPh3) (6)–(9) and [Ru2(CO)4(CH3CN)2(μ-L1)2] (10) and (11), where L1 is a 6-chloro- or 6-bromo-2-pyridonate ligand, are also efficient catalysts. Compared with catalyst 3, a halogen substituent at the pyridonate ligand affects the diastereoselectivity of cyclopropanation only slightly.Dinuclear Ru(I,I) complexes [Ru2(CO)4(μ-OOCR)2]n (R = CH3, C3H7, H, CF3) (1) and [Ru2(CO)4(μ-pyridin-2-olato)2]n (3) are efficient and effective catalysts for cyclopropanation of nucleophilic alkenes with methyl diazoacetate. The influence of these catalysts and related ones with 6-halopyridin-2-olate ligands is investigated.
Co-reporter:Martina Brunner;Frank-Gerrit Klärner
Helvetica Chimica Acta 2005 Volume 88(Issue 7) pp:1813-1825
Publication Date(Web):20 JUL 2005
DOI:10.1002/hlca.200590142
The cycloaddition of organic azides with some conjugated enamines of the 2-amino-1,3-diene, 1-amino-1,3-diene, and 2-aminobut-1-en-3-yne type is investigated. The 2-morpholinobuta-1,3-diene 1 undergoes regioselective [3+2] cycloaddition with several electrophilic azides RN32 (a, R=4-nitrophenyl; b, R=ethoxycarbonyl; c, R=tosyl; d, R=phenyl) to form 5-alkenyl-4,5-dihydro-5-morpholino-1H-1,2,3-triazoles 3 which are transformed into 1,5-disubstituted 1H-triazoles 4a,d or α,β-unsaturated carboximidamide 5 (Scheme 1). The cycloaddition reaction of 4-[(1E,3Z)-3-morpholino-4-phenylbuta-1,3-dienyl]morpholine (7) with azide 2a occurs at the less-substituted enamine function and yields the 4-(1-morpholino-2-phenylethenyl)-1H-1,2,3-triazole 8 (Scheme 2). The 1,3-dipolar cycloaddition reaction of azides 2a–d with 4-(1-methylene-3-phenylprop-2-ynyl)morpholine (9) is accelerated at high pressure (ca. 7–10 kbar) and gives 1,5-disubstituted dihydro-1H-triazoles 10a,b and 1-phenyl-5-(phenylethynyl)-1H-1,2,3-triazole (11d) in significantly improved yields (Schemes 3 and 4). The formation of 11d is also facilitated in the presence of an equimolar quantity of tBuOH. The three-component reaction between enamine 9, phenyl azide, and phenol affords the 5-(2-phenoxy-2-phenylethenyl)-1H-1,2,3-triazole 14d.

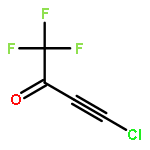
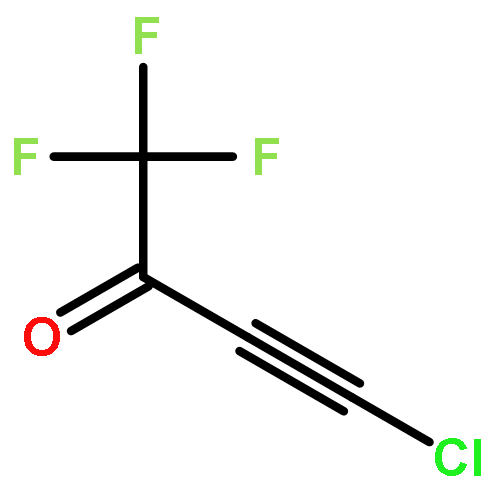
![METHANAMINIUM, N-[BIS(DIHEXYLAMINO)METHYLENE]-N-METHYL-, CHLORIDE](http://img.cochemist.com/ccimg/673000/672953-05-8.png)
![METHANAMINIUM, N-[BIS(DIHEXYLAMINO)METHYLENE]-N-METHYL-, CHLORIDE](http://img.cochemist.com/ccimg/673000/672953-05-8_b.png)
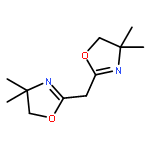
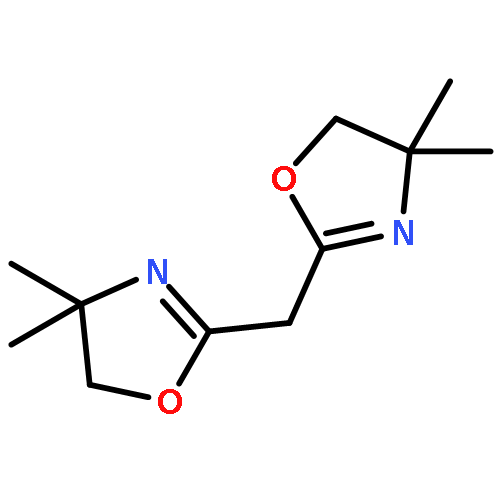

![2-Oxabicyclo[3.2.0]hept-6-ene-6,7-dicarboxylic acid, dimethyl ester](http://img.cochemist.com/ccimg/112200/112158-23-3.png)
![2-Oxabicyclo[3.2.0]hept-6-ene-6,7-dicarboxylic acid, dimethyl ester](http://img.cochemist.com/ccimg/112200/112158-23-3_b.png)
![1-HEXANAMINIUM, N-[BIS(DIMETHYLAMINO)METHYLENE]-N-HEXYL-, CHLORIDE](http://img.cochemist.com/ccimg/89700/89609-70-1.png)
![1-HEXANAMINIUM, N-[BIS(DIMETHYLAMINO)METHYLENE]-N-HEXYL-, CHLORIDE](http://img.cochemist.com/ccimg/89700/89609-70-1_b.png)




![Silane, (1,1-dimethylethyl)dimethyl[(2-methyl-1-cyclopenten-1-yl)oxy]-](http://img.cochemist.com/ccimg/68100/68081-17-4.png)
![Silane, (1,1-dimethylethyl)dimethyl[(2-methyl-1-cyclopenten-1-yl)oxy]-](http://img.cochemist.com/ccimg/68100/68081-17-4_b.png)


![2-Oxabicyclo[4.2.0]oct-7-ene-7,8-dicarboxylic acid, dimethyl ester, cis-](http://img.cochemist.com/ccimg/20500/20408-51-9.png)
![2-Oxabicyclo[4.2.0]oct-7-ene-7,8-dicarboxylic acid, dimethyl ester, cis-](http://img.cochemist.com/ccimg/20500/20408-51-9_b.png)
![Silane, (1,1-dimethylethyl)dimethyl[(2-methyl-1-cyclohexen-1-yl)oxy]-](http://img.cochemist.com/ccimg/20200/20152-33-4.png)
![Silane, (1,1-dimethylethyl)dimethyl[(2-methyl-1-cyclohexen-1-yl)oxy]-](http://img.cochemist.com/ccimg/20200/20152-33-4_b.png)