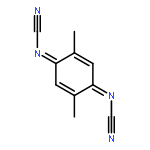
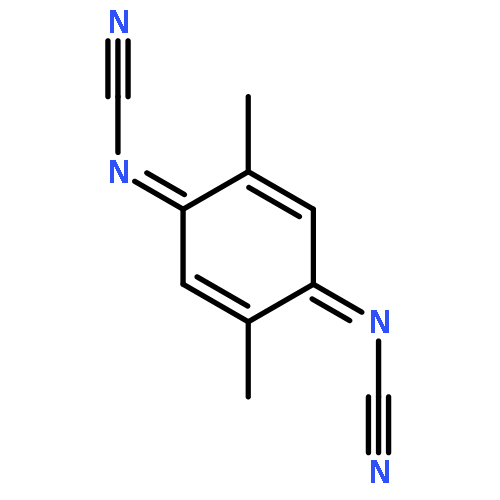
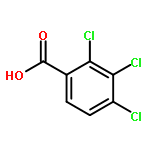
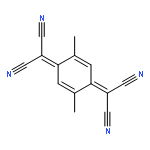
Co-reporter:Chihiro Kachi-Terajima, Rikako Ishii, Yoshiaki Tojo, Masato Fukuda, Yasutaka Kitagawa, Mizuki Asaoka, and Hitoshi Miyasaka
The Journal of Physical Chemistry C June 8, 2017 Volume 121(Issue 22) pp:12454-12454
Publication Date(Web):May 15, 2017
DOI:10.1021/acs.jpcc.7b03336
A series of MnIII saltmen dimers, [Mn2(5-Rsaltmen)2(X)2](A)2n (saltmen2– = N,N′-(1,1,2,2-tetramethylethylene)bis(salicylideneiminate); R = H, Cl, Br, MeO, Me; X = H2O, ReO4–, NO3–, N3–, NCS–, A– = ClO4–, PF6–, CF3SO3– for X = H2O) were synthesized and structurally and magnetically investigated to understand the correlation between their intradimer ferromagnetic (FM) interaction and single-molecule magnet (SMM) behavior. All complexes had a similar di-μ-phenolate-bridged out-of-plane dimer structure but displayed different bridging Mn–Oph* distances depending on the R substituents of the saltmen ligand and axial X ligand. Magnetic susceptibility studies revealed intradimer FM coupling (JMn–Mn*), resulting in an ST = 4 ground state for all dimers. However, the magnitude of FM coupling strongly depended on R and X. JMn–Mn* increased with decreasing Mn–Oph* distance but decreased with decreasing Mn–X distance with a relation of H2O ≈ ReO4– > NO3– > N3– ≈ NCS– with a linear trend for R = H, Cl, Me but not for R = Br, MeO. Theoretical investigations revealed that a larger orbital overlap stabilized a FM spin configuration through competition between the orbital degeneracy and on-site Coulomb repulsion of out-of-phase and in-phase orbitals. Most dimers showed typical SMM behavior. The dimers with larger JMn–Mn* tended to have higher blocking temperatures.
Co-reporter:Yoshihiro Sekine;Masanori Tonouchi;Taiga Yokoyama;Wataru Kosaka
CrystEngComm (1999-Present) 2017 vol. 19(Issue 17) pp:2300-2304
Publication Date(Web):2017/05/02
DOI:10.1039/C7CE00492C
A TTF–TCNQ charge-transfer salt was incorporated in a layer structure with paddlewheel-type carboxylate-bridged dirhodium(II, II) complexes ([Rh2]) to construct π-stacked pillared layer frameworks of formula (TTF)n[{Rh}2(TCNQ)] (n = 1 or 2).
Co-reporter:Jun Zhang, Wataru Kosaka, Hiroki Fukunaga, Susumu Kitagawa, Masaki Takata, and Hitoshi Miyasaka
Inorganic Chemistry 2016 Volume 55(Issue 22) pp:12085
Publication Date(Web):November 7, 2016
DOI:10.1021/acs.inorgchem.6b02349
On-demand design of porous frameworks for selective capture of specific gas molecules, including toxic gas molecules such as nitric oxide (NO), is a very important theme in the research field of molecular porous materials. Herein, we report the achievement of highly selective NO adsorption through chemical doping in a framework (i.e., solid solution approach): the highly electron donating unit [Ru2(o-OMePhCO2)4] (o-OMePhCO2– = o-anisate) was transplanted into the structurally flexible chain framework [Ru2(4-Cl-2-OMePhCO2)4(phz)] (0; 4-Cl-2-OMePhCO2– = 4-chloro-o-anisate and phz = phenazine) to obtain a series of doped compounds, [{Ru2(4-Cl-2-OMePhCO2)4}1–x{Ru2(o-OMePhCO2)4}x(phz)] (x = 0.34, 0.44, 0.52, 0.70, 0.81, 0.87), with [Ru2(o-OMePhCO2)4(phz)] (1) as x = 1. The original compound 1 was made purely from a “highly electron donating unit” but had no adsorption capability for gases because of its nonporosity. Meanwhile, the partial transplant of the electronically advantageous [Ru2(o-OMePhCO2)4] unit with x = 0.34–0.52 in 0 successfully enhanced the selective adsorption capability of NO in an identical structurally flexible framework; an uptake at 95 kPa that was 1.7–3 mol/[Ru2] unit higher than that of the original 0 compound was achieved (121 K). The solid solution approach is an efficient means of designing purposeful porous frameworks.
Co-reporter:Keita Nakabayashi, Masaki Nishio, and Hitoshi Miyasaka
Inorganic Chemistry 2016 Volume 55(Issue 5) pp:2473-2480
Publication Date(Web):February 15, 2016
DOI:10.1021/acs.inorgchem.5b02858
The stepwise neutral–ionic (N–I) phase transition found in the alternating donor/acceptor (DA) chain [Ru2(2,3,5,6-F4PhCO2)4(DMDCNQI)]·2(p-xylene) (0; 2,3,5,6-F4PhCO2– = 2,3,5,6-tetrafluorobenzoate; DMDCNQI = 2,5-dimethyl-N,N′-dicyanoquinonediimine) was tuned by partly substituting the acceptor DMDCNQI with 2,5-dimethoxy-N,N′-dicyanoquinonediimine (DMeODCNQI), which displays a poorer electron affinity in an isostructural series. The site-doped series comprised [Ru2(2,3,5,6-F4PhCO2)4(DMDCNQI)1–x(DMeODCNQI)x]·2(p-xylene) for doping rates (x) = 0.05 (0.05-MeO), 0.10 (0.10-MeO), 0.15 (0.15-MeO), and 0.20 (0.20-MeO). The neutral chain [Ru2(2,3,5,6-F4PhCO2)4(DMeODCNQI)]·4(p-xylene) (1), which only contained DMeODCNQI, was also characterized. All site-doped compounds were isostructural to 0 except 1 despite their identical DA chain motif. Except at an x value of 0.20, they displayed a two-step N–I transition involving an intermediate phase. This transition occurred at high temperatures in 0 but shifted to lower temperatures in a parallel manner with increasing doping rate. Simultaneously, each transition broadened with increasing doping rate, leading to a convergence of two transitions at an x value approximating 0.2. Donor/acceptor-site-doping techniques present somewhat different impacts in terms of interchain Coulomb effects.
Co-reporter:Yoshihiro Sekine, Wataru Kosaka, Hirohisa Kano, Changxiao Dou, Taiga Yokoyama and Hitoshi Miyasaka
Dalton Transactions 2016 vol. 45(Issue 17) pp:7427-7434
Publication Date(Web):14 Mar 2016
DOI:10.1039/C6DT00569A
Carboxylate-ligand substitution reactions of paddlewheel-type diruthenium(II, III) complexes ([RuII,III2(RCO2)4]+) with 2,6-bis(trifluoromethyl)benzoate (2,6-(CF3)2PhCO2−) involving a selective reduction to [RuII,II2] provide a series of trans-substituted paddlewheel-type diruthenium(II, II) complexes, [RuII,II2(2,6-(CF3)2PhCO2)2(RCO2)2(THF)2] (R = CH3, 1; C2H5, 2; C3H7, 3; C4H9, 4; C(CH3)3, 5; 2,3,5,6-F4Ph, 6). Crystal structures of 1–6 were determined, and their electronic states were investigated by cyclic voltammetry, density functional theory (DFT) and magnetic measurements. This is the first example of trans-heteroleptic carboxylate-bridged [RuII,II2] complexes.
Co-reporter:Hiroki Fukunaga, Takafumi Yoshino, Hajime Sagayama, Jun-ichi Yamaura, Taka-hisa Arima, Wataru Kosaka and Hitoshi Miyasaka
Chemical Communications 2015 vol. 51(Issue 37) pp:7795-7798
Publication Date(Web):17 Mar 2015
DOI:10.1039/C5CC01633A
A novel charge-disproportionation state with δ = 0.75 was observed in an electron-donor (D)/-acceptor (A) Dδ+2A2δ− layered framework by chemically tuning the electron-donating affinity of D at the boundary between D0.5+2A− and D+2A2− phases, which was pressure-sensitive due to the formation of the D+2A2− oxidation state.
Co-reporter:Wataru Kosaka, Hiroki Fukunaga, and Hitoshi Miyasaka
Inorganic Chemistry 2015 Volume 54(Issue 20) pp:10001-10006
Publication Date(Web):September 28, 2015
DOI:10.1021/acs.inorgchem.5b01776
The donor (D)/acceptor (A) assembly reaction of the paddlewheel-type diruthenium(II,II) complex [Ru2(2,4,6-F3PhCO2)4(THF)2] (2,4,6-F3PhCO2– = 2,4,6-trifluorobenzoate; abbreviated hereafter as [Ru2]) with 7,7,8,8-tetracyano-p-quinodimethane (TCNQ) in a p-xylene/CH2Cl2 solvent system led to the formation of a two-dimensional layered compound, [{Ru2(2,4,6-F3PhCO2)4}2(TCNQ)]·2(p-xylene)·2CH2Cl2 (1). As expected from this D/A combination, 1 has a one-electron-transfer ionic state with the D0.5+2A– formulation. This state formally derives a heterospin state composed of S = 1 for [RuII,II2], S = 3/2 for [RuII,III2]+, and S = 1/2 for TCNQ•–, possibly causing intralayer ferrimagnetic spin ordering. Most of these types of compounds have an antiferromagnetic ground state because of the coupling of ferrimagnetically ordered layers in dipole antiferromagnetic interactions. However, 1 became a three-dimensional ferrimagnet with TC = 91 K because of the presence of interlayer ferromagnetic interactions.
Co-reporter:Wataru Kosaka, Takaumi Morita, Taiga Yokoyama, Jun Zhang, and Hitoshi Miyasaka
Inorganic Chemistry 2015 Volume 54(Issue 4) pp:1518-1527
Publication Date(Web):January 28, 2015
DOI:10.1021/ic502513p
In a series of two-dimensional layered frameworks constructed by two electron-donor (D) and one electron-acceptor (A) units (a D2A framework), two-electron transferred systems with D+2A2– were first synthesized as [{Ru2(R-PhCO2)4}2(TCNQRx)]·n(solv) (R = o-CF3, Rx = H2 (1), R = o-CF3, Rx = Me2 (2), R = o-CF3, Rx = F4 (3), R = o-Me, TCNQRx = BTDA-TCNQ (4), R = p-Me, TCNQRx = BTDA-TCNQ (5), where TCNQ is 7,7,8,8-tetracyano-p-quinodimethane and BTDA-TCNQ is bis[1,2,5]dithiazolotetracyanoquinodimethane). The D+2A2– system was synthesized by assembling D/A combinations of paddlewheel-type [Ru2II,II(R-PhCO2)4] complexes and TCNQRx that possibly caused a large gap between the HOMO of D and the LUMO of A (ΔEH–L(DA)). All compounds were paramagnetic because of quasi-isolated [Ru2II,III]+ units with weakly antiferromagnetically coupled S = 3/2 spins via diamagnetic TCNQRx2– and/or through the interlayer space. The ionic states of these compounds were determined using the HOMO/LUMO energies and redox potentials of the D and A components in the ionization diagram for ΔEH–L(DA) vs ΔE1/2(DA) (= E1/2(D) – E1/2(A); E1/2 = first redox potential) as well as by previously reported data for the D2A and DA series of [Ru2]/TCNQ, DCNQI materials. The boundary between the one-electron and the two-electron transferred ionic regimes (1e–I and 2e–I, respectively) was not characterized. Therefore, another diagram for ΔEH–L(DA) vs |2E1/2(A) – 1E1/2(A)|, where 2E1/2(A) and 1E1/2(A) are the second and first redox potentials of TCNQRx, respectively, was used because the 2e–I regime is dependent on on-site Coulomb repulsion (U = |2E1/2(A) – 1E1/2(A)|) of TCNQRx. This explained the oxidation states of 1–5 and the relationship between ΔEH–L(DA) and U and allowed us to determine whether the ionic regime was 1e–I or 2e–I. These diagrams confirm that a charge-oriented choice of building units is possible even when designing covalently bonded D2A framework systems.
Co-reporter:Wataru Kosaka, Masahisa Itoh and Hitoshi Miyasaka
Dalton Transactions 2015 vol. 44(Issue 17) pp:8156-8168
Publication Date(Web):20 Mar 2015
DOI:10.1039/C5DT00505A
A series of paddlewheel diruthenium(II, II) complexes with various chlorine-substituted benzoate ligands (Cl-series) was synthesized as tetrahydrofuran (THF) adducts [Ru2(ClxPhCO2)4(THF)2]; where ClxPhCO2− = o-chlorobenzoate, o-Cl; m-chlorobenzoate, m-Cl; p-chlorobenzoate, p-Cl; 2,3-dichlorobenzoate, 2,3-Cl2; 2,4-dichlorobenzoate, 2,4-Cl2; 2,5-dichlorobenzoate, 2,5-Cl2; 2,6-dichlorobenzoate, 2,6-Cl2; 3,4-dichlorobenzoate, 3,4-Cl2; 3,5-dichlorobenzoate, 3,5-Cl2; 2,3,4-trichlorobenzoate, 2,3,4-Cl3; 2,3,5-trichlorobenzoate, 2,3,5-Cl3; 2,4,5-trichlorobenzoate, 2,4,5-Cl3; 3,4,5-trichlorobenzoate, 3,4,5-Cl3; 2,3,4,5-tetrachlorobenzoate, 2,3,4,5-Cl4. This Cl-series and the previously synthesized F-series together with four new fluorine-substituted derivatives, [Ru2(FxPhCO2)4(THF)2] (where FxPhCO2− = 2,3-difluorobenzoate, 2,3-F2; 2,4-difluorobenzoate, 2,4-F2; 2,5-difluorobenzoate, 2,5-F2; 2,3,5-trifluorobenzoate, 2,3,5-F3), were experimentally characterized with respect to solid-state structure, magnetic properties and electrochemistry. By tuning the substituents of the benzoate ligands using chlorine or fluorine atoms, the redox potential (E1/2) for [Ru2II,II]/[Ru2II,III]+ varied over a wide range of potentials from −40 mV to 360 mV (vs. Ag/Ag+ in THF). This was dependent on (i) the number of ortho-substituents, i.e. non-, mono- and di-o-substituted groups, with quasi-Hammett parameters for ortho-Cl and -F substitutions (σo = −0.272 and −0.217, respectively) and (ii) the general Hammett constants, σm and σp, for each group. The HOMO energy level calculated on the basis of the atomic coordinates of the solid-state structure was strongly affected by Cl- and F-substitutions as well as the redox potential in solution, which emphasizes the steric contribution of ortho-substituents in the energy level giving a deviation of EHOMO < 0.3 eV and <0.55 eV for the Cl- and F-series, respectively.
Co-reporter:Masaki Nishio, Natsuko Motokawa and Hitoshi Miyasaka
CrystEngComm 2015 vol. 17(Issue 40) pp:7618-7622
Publication Date(Web):24 Jul 2015
DOI:10.1039/C5CE01260K
Fishnet-like layered compounds comprising trifluoroacetate-bridged paddlewheel dimetal(II, II) complexes and 7,7,8,8-tetracyano-p-quinodimethane (TCNQ) were crystalized with anthracene (ANT) molecules between their layers and crystallization solvents at their hexagonal columnar pores. These complexes undergo a solvent-release crystal-to-crystal transformation involving the migration of a portion of the ANT molecules from between the layers to the hexagonal pores, effectively locking the slidable layers.
Co-reporter:Hiroki Fukunaga;Dr. Hitoshi Miyasaka
Angewandte Chemie International Edition 2015 Volume 54( Issue 2) pp:569-573
Publication Date(Web):
DOI:10.1002/anie.201410057
Abstract
The control of inter-lattice magnetic interactions is a crucial issue when long-range ordered magnets that are based on low-dimensional magnetic frameworks are designed. A “pillared layer framework (PLF)” model could be an efficient system for this purpose. In this report, A magnet based on a π-stacked PLF with a phase transition temperature of 82 K, which can be increased to 107 K by applying a pressure of 12.5 kbar, is rationally constructed. Two types of low-dimensional magnetic framework systems, an electron donor/acceptor magnetic layer and a charge transfer [FeCp*2]+TCNQ.− columnar magnet ([FeCp*2]+=decamethylferrocenium; TCNQ=7,7,8,8-tetracyano-p-quinodimethane), are integrated to fabricate the magnet. This synthetic strategy employing a combination of layers and chains is widely useful not only for magnet design, but also for the creation of multifunctional materials with pores and anisotropic frameworks.
Co-reporter:Hiroki Fukunaga;Dr. Hitoshi Miyasaka
Angewandte Chemie 2015 Volume 127( Issue 2) pp:579-583
Publication Date(Web):
DOI:10.1002/ange.201410057
Abstract
The control of inter-lattice magnetic interactions is a crucial issue when long-range ordered magnets that are based on low-dimensional magnetic frameworks are designed. A “pillared layer framework (PLF)” model could be an efficient system for this purpose. In this report, A magnet based on a π-stacked PLF with a phase transition temperature of 82 K, which can be increased to 107 K by applying a pressure of 12.5 kbar, is rationally constructed. Two types of low-dimensional magnetic framework systems, an electron donor/acceptor magnetic layer and a charge transfer [FeCp*2]+TCNQ.− columnar magnet ([FeCp*2]+=decamethylferrocenium; TCNQ=7,7,8,8-tetracyano-p-quinodimethane), are integrated to fabricate the magnet. This synthetic strategy employing a combination of layers and chains is widely useful not only for magnet design, but also for the creation of multifunctional materials with pores and anisotropic frameworks.
Co-reporter:Wataru Kosaka ; Kayo Yamagishi ; Jun Zhang
Journal of the American Chemical Society 2014 Volume 136(Issue 35) pp:12304-12313
Publication Date(Web):August 13, 2014
DOI:10.1021/ja504992g
The gate-opening adsorption behavior of the one-dimensional chain compound [Ru2(4-Cl-2-OMePhCO2)4(phz)] (1; 4-Cl-2-OMePhCO2– = 4-chloro-o-anisate; phz = phenazine) for various gases (O2, NO, and CO2) was electronically monitored in situ by applying ac electric fields to pelletized samples attached to a cryostat, which was used to accurately control the temperature and gas pressure. The gate-opening and -closing transitions induced by gas adsorption/desorption, respectively, were accurately monitored by a sudden change in the real part of permittivity (ε′). The transition temperature (TGO) was also found to be dependent on the applied temperature and gas pressure according to the Clausius–Clapeyron equation. This behavior was also observed in the isostructural compound [Rh2(4-Cl-2-OMePhCO2)4(phz)] (2), which exhibited similar gate-opening adsorption properties, but was not detected in the nonporous gate-inactive compound [Ru2(o-OMePhCO2)4(phz)] (3). Furthermore, the imaginary part of permittivity (ε″) effectively captured the electronic perturbations of the samples induced by the introduced guest molecules. Only the introduction of NO resulted in the increase of the sample’s electronic conductivity for 1 and 3, but not for 2. This behavior indicates that electronic host–guest interactions were present, albeit very weak, at the surface of sample 1 and 3, i.e., through grain boundaries of the sample, which resulted in perturbation of the conduction band of this material’s framework. This technique involving the in situ application of ac electric fields is useful not only for rapidly monitoring gas sorption responses accompanied by gate-opening/-closing structural transitions but also potentially for the development of molecular framework materials as chemically driven electronic devices.
Co-reporter:Masaki Nishio and Hitoshi Miyasaka
Inorganic Chemistry 2014 Volume 53(Issue 9) pp:4716-4723
Publication Date(Web):April 21, 2014
DOI:10.1021/ic500413j
The donor/acceptor ionic chain (i.e., the D+A– chain) [Ru2(2-MeO-4-ClPhCO2)4(BTDA-TCNQ)]·2.5(benzene) (1; 2-MeO-4-ClPhCO2– = 2-methoxy-4-chlorobenzoate; BTDA-TCNQ = bis(1,2,5-thiadiazolo)tetracyanoquinodimethane) is a ferrimagnetic chain with S = 3/2 from [Ru2II,III]+ (i.e., D+) and S = 1/2 from BTDA-TCNQ•– (i.e., A–), with J ≈ −100 K, in which long-range antiferromagnetic ordering at TN = 11 K occurs because interchain antiferromagnetic interactions are critical. Compound 1 undergoes a reversible crystal-to-crystal structural transformation with the elimination/absorption of the crystallization solvent to form the dried compound [Ru2(2-MeO-4-ClPhCO2)4(BTDA-TCNQ)] (1′), which has a higher TN (14 K). This change is clearly caused by the shortening of the interchain distances because the exchange coupling parameter for the chain is the same in both 1 and 1′. The chain compounds in 1 can be doped with minor diamagnetic [Rh2II,II] species, [{(Ru2)1–x(Rh2)x(2-MeO-4-ClPhCO2)4}(BTDA-TCNQ)]·2.5(benzene) (x = 0.03 for Rh-3%; x = 0.05 for Rh-5%; x = 0.16 for Rh-16%), which shifts the TN to lower temperatures, the magnitude of the shift being dependent on the doping ratio x (TN = 5.9 K for Rh-3%, TN = 3.7 K for Rh-5%, and TN was not observed above 1.8 K for Rh-16%). Drying a doped compound increased its TN, as was found for 1′: TN = 9.9 K for Rh-3%′, TN = 9.2 K for Rh-5%′, and TN was not observed above 1.8 K for Rh-16%′. TN had a linear relationship with the doping ratio x of the [Rh2] species in both the fresh and dried compounds. The TN linear relationship is associated with the magnitude of the effective magnetic dipole (i.e., the average correlation length) in the chains caused by the [Rh2] defects as well as naturally generated defects in the synthetic process and with the interchain distances affected by the crystal-to-crystal transformations. These results demonstrate that slightly modifying the short-range correlation lengths, which changes the magnetic dipole magnitudes, strongly affects the bulk antiferromagnetic transition, with key dipole–dipole interactions, in low-dimensional anisotropic systems.
Co-reporter:Keita Nakabayashi;Dr. Hitoshi Miyasaka
Chemistry - A European Journal 2014 Volume 20( Issue 17) pp:5121-5131
Publication Date(Web):
DOI:10.1002/chem.201304420
Abstract
The temperature-induced stepwise neutral–ionic (N–I) phase transition in the covalently bonded donor–acceptor chain compound [Ru2(2,3,5,6-F4PhCO2)4DMDCNQI]⋅ 2(p-xylene) (2,3,5,6-F4PhCO2−=2,3,5,6-tetrafluorobenzoate; DMDCNQI=2,5-dimethyl-N,N′-dicyanoquinodiimine) was systematically tuned over a wide temperature range using two techniques: 1) A chemical technique based on doping with a less-active donor unit [Ru2II,II(F5PhCO2)4] (F5PhCO2−=pentafluorobenzoate), thereby providing an isostructural doped series [{Ru2II,II(2,3,5,6-F4PhCO2)4}1−x{Ru2II,II(F5PhCO2)4}xDMDCNQI]⋅2(p-xylene), with x=0.06, 0.10, 0.21, and 0.24; and 2) a physical technique, which was the application of hydrostatic pressure to the doped compounds. The stepwise N–I transition observed in the original compound was systematically varied in terms of the viewpoints of both transition temperature and transition features (stepwise or monotonic) dependent on the amount of dopants x. Application of pressure efficiently tuned the N–I transitions, with the oxidation phases being dramatically modified by applying only weak pressure up to 4 kbar. Even in cases that led to N–I transitions in small domains of the chains at ambient pressure, the application of pressure caused an expansion of the domains that enabled N–I transitions, finally leading to a complete change in the oxidation state of the chains, from neutral to ionic, accompanied by a change from a paramagnetic state to a ferrimagnetically ordered state.
Co-reporter:Masaki Nishio ; Norihisa Hoshino ; Wataru Kosaka ; Tomoyuki Akutagawa
Journal of the American Chemical Society 2013 Volume 135(Issue 47) pp:17715-17718
Publication Date(Web):October 28, 2013
DOI:10.1021/ja409785a
On the basis of the concept that the design of a mixed valence system is a key route to create electronic conducting frameworks, we propose a unique idea to rationally produce mixed valency in an ionic donor/acceptor chain (i.e., D+A– chain). The doping of a redox-inert (insulator) dopant (P) into a D+A– chain in place of neutral D enables the creation of mixed valency A0/A– domains between P units: P–(D+A–)nA0–P, where n is directly dependent on the dopant ratio, and charge transfer through the P units leads to electron transport along the framework. This hypothesis was experimentally demonstrated in an ionic DA chain synthesized from a redox-active paddlewheel [Ru2II,II] complex and TCNQ derivative by doping with a redox-inert [Rh2II,II] complex.
Co-reporter:Wataru Kosaka ; Kayo Yamagishi ; Akihiro Hori ; Hiroshi Sato ; Ryotaro Matsuda ; Susumu Kitagawa ; Masaki Takata
Journal of the American Chemical Society 2013 Volume 135(Issue 49) pp:18469-18480
Publication Date(Web):October 23, 2013
DOI:10.1021/ja4076056
The design of porous materials that undergo selective adsorption of a specific molecule is a critical issue in research on porous coordination polymers or metal–organic frameworks. For the purpose of the selective capture of molecules possessing an electron-acceptor character such as nitric oxide (NO), one-dimensional chain compounds possessing a high donor character have been synthesized using 4-chloroanisate-bridged paddlewheel-type dimetal(II, II) complexes with M = Ru and Rh and phenazine (phz) as the chain linker: [M2(4-Cl-2-OMePhCO2)4(phz)]·n(CH2Cl2) (M = Ru, 1; Rh, 2). These compounds are isostructural and are composed of chains with a [−{M2}–phz−] repeating unit and CH2Cl2 occupying the void space between the chains. Compounds 1 and 2 change to a new phase (1-dry and 2-dry) upon evacuating the crystallization solvent (CH2Cl2) and almost lose their pores in the drying process: no void space in 1-dry and 31.8 Å3, corresponding to 2.9% of the cell volume, in 2-dry. Nevertheless, the compounds show a unique gas accommodation ability. Accompanied by a structural transformation (i.e., the first gate-opening) at low pressures of <10 kPa, both compounds show a typical physisorption isotherm for O2 (90 K) and CO2 (195 K), with the adsorption amount of ca. 2–4 gas molecules per [M2] unit. In addition, the adsorption isotherm for NO (121 K) involves the first gate-opening followed by a second gate-opening anomaly at NO pressures of ≈52 kPa for 1-dry and ≈21 kPa for 2-dry. At the first gate-opening, the absorbed amount of NO is ca. 4 molecules per [M2] unit, and then it reaches 8.4 and 6.3 for 1-dry and 2-dry, respectively, at 95 kPa. Only the isotherm for NO exhibits hysteresis in the desorption process, and some of the NO molecules are trapped in pores even after evacuating at 121 K, although it recovers to the original dried sample on heating to room temperature. The adsorbed NO molecules accrue a significant electron donation from the host framework even in the [Rh2] derivative, indicating that such simple porous compounds with electron-donor characteristics are useful for the selective adsorption of NO.
Co-reporter:Wataru Kosaka, Naoto Yamamoto, and Hitoshi Miyasaka
Inorganic Chemistry 2013 Volume 52(Issue 17) pp:9908-9914
Publication Date(Web):August 20, 2013
DOI:10.1021/ic401030r
The reactions of paddlewheel-type diruthenium(II, II) complexes, [Ru2II,II(x-FPhCO2)4(THF)2] (x-FPhCO2– = x-fluorobenzoate with x- = o-, m-, p-), with 2,6-diaminopyridine (dapy) and 7-azaindole (azain) afford axially capped discrete compounds, [Ru2II,II(x-FPhCO2)4(dapy)2] (x = o-, 1; m-, 2; p-, 3) and [Ru2II,II(o-FPhCO2)4(azain)2] (4), respectively. In these compounds, intramolecular hydrogen bonds are observed between NH2 groups for 1–3 or imine NH groups for 4 and oxygen atoms of carboxylate groups. In addition, hydrogen bonds of NH2···F are also observed for 1 and 4 with an o-positioned F atom on benzoate. This coordination mode, i.e., a dual bonding mode with σ-bonding and hydrogen bonding, should assist ligand coordination to the axial position of the [Ru2] unit. The Ru–N bond distance in 1–4 is shorter than that observed in related compounds reported previously. In a similar fashion, reactions with planar MII dithiobiuret (dtb) complexes, [MII(dtb)2] (MII = PdII and PtII), were carried out. One-dimensional alternating chains, [{Ru2II,II(o-FPhCO2)4}{MII(dtb)2}] (MII = PdII, 5; PtII, 6), were obtained, in which the hydrogen-bonding modes of NH2···O and NH2···F are present, as expected. DFT calculations for the [MII(dtb)2] unit revealed that the LUMO of [MII(dtb)2] lies at −2.159 and −1.781 eV for M = Pd and Pt, respectively, which is much higher than HOMO energy at −4.184 eV calculated for [Ru2II,II(o-FPhCO2)(THF)2], proving that the respective units are essentially electronically isolated in the chains.
Co-reporter:Masaki Nishio, Natsuko Motokawa, Miho Takemura and Hitoshi Miyasaka
Dalton Transactions 2013 vol. 42(Issue 45) pp:15898-15901
Publication Date(Web):11 Jul 2013
DOI:10.1039/C3DT51271A
Pyrene-intercalated layered compounds, [{Ru2(O2CCF3)4}2(TCNQRx)]·2(pyrene) (TCNQRx = 7,7,8,8-tetracyano-p-quinodimethane derivatives; Rx = H4 and F4), were synthesized. Pyrene prohibits intralayer electron transfer of [Ru2II,II] → TCNQRx, even in the compound with Rx = F4, from occurring.
Co-reporter:Akihiro Hashikawa, Yuki Sawada, Yuma Yamamoto, Masaki Nishio, Wataru Kosaka, Yoshihito Hayashi and Hitoshi Miyasaka
CrystEngComm 2013 vol. 15(Issue 24) pp:4852-4859
Publication Date(Web):11 Apr 2013
DOI:10.1039/C3CE40426A
The assembly reactions of a pivalate-bridged paddlewheel diruthenium(II, III) complex, [Ru2II,III(piv)4(THF)2]BF4 (piv = pivalate), with three types of polyoxometalate (POM) clusters, (TBA)2[Mo6O19] (TBA = tetra-n-butylammonium cation; Lindqvist-type Mo6 POM), (TBA)3[H3V10O28] (V10-type POM), and (THA)3[PMo12O40] (THA = tetra-n-hexylammonium cation; Keggin-type Mo12 POM), led successfully to the formation of one-, two-, and three-dimensional (1-, 2-, and 3-D) frameworks composed of [–{Ru2}–POM–] inorganic linkages in (TBA)[{Ru2(piv)4}{Mo6O19}]·CH2Cl2·C2H4Cl2 (1), (TBA)[{Ru2(piv)4}2{H3V10O28}]·C2H4Cl2·7H2O (2), and (THA)[{Ru2(piv)4}2{PMo12O40}]·2(C2H4Cl2) (3), respectively, where 1 is a 1-D linear chain, 2 forms a layered 2-D sheet framework, and 3 constructs a cristobalite-type 3-D structure.
Co-reporter:Yoshihiro Sekine, Taiga Yokoyama, Norihisa Hoshino, Manabu Ishizaki, Katsuhiko Kanaizuka, Tomoyuki Akutagawa, Masa-aki Haga and Hitoshi Miyasaka
Chemical Communications 2016 - vol. 52(Issue 97) pp:NaN13986-13986
Publication Date(Web):2016/11/08
DOI:10.1039/C6CC08310B
Novel thin films composed of a donor (D)/acceptor (A) charge-transfer chain compound were fabricated by a layer-by-layer technique using complexation of a paddlewheel-type diruthenium(II, II) complex with an N,N′-dicyanoquinonediimine derivative on an ITO substrate with a pyridine-substituted phosphonate anchor. The stepwise growth of an electron-transfer D+A−-chain thin film was confirmed.
Co-reporter:Hiroki Fukunaga, Takafumi Yoshino, Hajime Sagayama, Jun-ichi Yamaura, Taka-hisa Arima, Wataru Kosaka and Hitoshi Miyasaka
Chemical Communications 2015 - vol. 51(Issue 37) pp:NaN7798-7798
Publication Date(Web):2015/03/17
DOI:10.1039/C5CC01633A
A novel charge-disproportionation state with δ = 0.75 was observed in an electron-donor (D)/-acceptor (A) Dδ+2A2δ− layered framework by chemically tuning the electron-donating affinity of D at the boundary between D0.5+2A− and D+2A2− phases, which was pressure-sensitive due to the formation of the D+2A2− oxidation state.
Co-reporter:Yoshihiro Sekine, Wataru Kosaka, Hirohisa Kano, Changxiao Dou, Taiga Yokoyama and Hitoshi Miyasaka
Dalton Transactions 2016 - vol. 45(Issue 17) pp:NaN7434-7434
Publication Date(Web):2016/03/14
DOI:10.1039/C6DT00569A
Carboxylate-ligand substitution reactions of paddlewheel-type diruthenium(II, III) complexes ([RuII,III2(RCO2)4]+) with 2,6-bis(trifluoromethyl)benzoate (2,6-(CF3)2PhCO2−) involving a selective reduction to [RuII,II2] provide a series of trans-substituted paddlewheel-type diruthenium(II, II) complexes, [RuII,II2(2,6-(CF3)2PhCO2)2(RCO2)2(THF)2] (R = CH3, 1; C2H5, 2; C3H7, 3; C4H9, 4; C(CH3)3, 5; 2,3,5,6-F4Ph, 6). Crystal structures of 1–6 were determined, and their electronic states were investigated by cyclic voltammetry, density functional theory (DFT) and magnetic measurements. This is the first example of trans-heteroleptic carboxylate-bridged [RuII,II2] complexes.
Co-reporter:Masaki Nishio, Natsuko Motokawa, Miho Takemura and Hitoshi Miyasaka
Dalton Transactions 2013 - vol. 42(Issue 45) pp:NaN15901-15901
Publication Date(Web):2013/07/11
DOI:10.1039/C3DT51271A
Pyrene-intercalated layered compounds, [{Ru2(O2CCF3)4}2(TCNQRx)]·2(pyrene) (TCNQRx = 7,7,8,8-tetracyano-p-quinodimethane derivatives; Rx = H4 and F4), were synthesized. Pyrene prohibits intralayer electron transfer of [Ru2II,II] → TCNQRx, even in the compound with Rx = F4, from occurring.
Co-reporter:Wataru Kosaka, Masahisa Itoh and Hitoshi Miyasaka
Dalton Transactions 2015 - vol. 44(Issue 17) pp:NaN8168-8168
Publication Date(Web):2015/03/20
DOI:10.1039/C5DT00505A
A series of paddlewheel diruthenium(II, II) complexes with various chlorine-substituted benzoate ligands (Cl-series) was synthesized as tetrahydrofuran (THF) adducts [Ru2(ClxPhCO2)4(THF)2]; where ClxPhCO2− = o-chlorobenzoate, o-Cl; m-chlorobenzoate, m-Cl; p-chlorobenzoate, p-Cl; 2,3-dichlorobenzoate, 2,3-Cl2; 2,4-dichlorobenzoate, 2,4-Cl2; 2,5-dichlorobenzoate, 2,5-Cl2; 2,6-dichlorobenzoate, 2,6-Cl2; 3,4-dichlorobenzoate, 3,4-Cl2; 3,5-dichlorobenzoate, 3,5-Cl2; 2,3,4-trichlorobenzoate, 2,3,4-Cl3; 2,3,5-trichlorobenzoate, 2,3,5-Cl3; 2,4,5-trichlorobenzoate, 2,4,5-Cl3; 3,4,5-trichlorobenzoate, 3,4,5-Cl3; 2,3,4,5-tetrachlorobenzoate, 2,3,4,5-Cl4. This Cl-series and the previously synthesized F-series together with four new fluorine-substituted derivatives, [Ru2(FxPhCO2)4(THF)2] (where FxPhCO2− = 2,3-difluorobenzoate, 2,3-F2; 2,4-difluorobenzoate, 2,4-F2; 2,5-difluorobenzoate, 2,5-F2; 2,3,5-trifluorobenzoate, 2,3,5-F3), were experimentally characterized with respect to solid-state structure, magnetic properties and electrochemistry. By tuning the substituents of the benzoate ligands using chlorine or fluorine atoms, the redox potential (E1/2) for [Ru2II,II]/[Ru2II,III]+ varied over a wide range of potentials from −40 mV to 360 mV (vs. Ag/Ag+ in THF). This was dependent on (i) the number of ortho-substituents, i.e. non-, mono- and di-o-substituted groups, with quasi-Hammett parameters for ortho-Cl and -F substitutions (σo = −0.272 and −0.217, respectively) and (ii) the general Hammett constants, σm and σp, for each group. The HOMO energy level calculated on the basis of the atomic coordinates of the solid-state structure was strongly affected by Cl- and F-substitutions as well as the redox potential in solution, which emphasizes the steric contribution of ortho-substituents in the energy level giving a deviation of EHOMO < 0.3 eV and <0.55 eV for the Cl- and F-series, respectively.


![Ruthenium, tetrakis[m-(acetato-kO:kO')]chlorodi-, (Ru-Ru)](http://img.cochemist.com/ccimg/38900/38833-34-0.png)
![Ruthenium, tetrakis[m-(acetato-kO:kO')]chlorodi-, (Ru-Ru)](http://img.cochemist.com/ccimg/38900/38833-34-0_b.png)