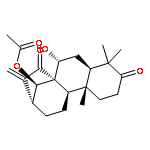
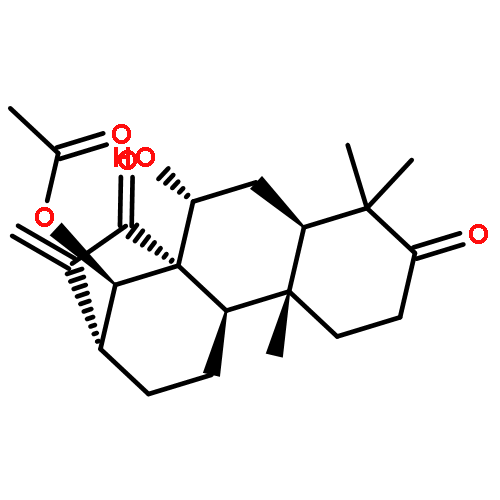
Co-reporter:Tian-Ya Liu;Xiao-Ying Yang;Long-Tai Zheng;Guang-Hui Wang;Xue-Chu Zhen
Journal of Neurochemistry 2017 Volume 140(Issue 4) pp:589-604
Publication Date(Web):2017/02/01
DOI:10.1111/jnc.13907
AbstractMicroglia-mediated neuroinflammation plays a critical role in the pathological development of Parkinson's disease (PD). Orphan nuclear receptor Nur77 (Nur77) is abundant in neurons, while its role in microglia-mediated neuroinflammation remains unclear. The present data demonstrated that the expression of Nur77 in microglia was reduced accompanied by microglia activation in response to lipopolysaccharide (LPS) in vitro and in experimental 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-PD mouse model. Nur77 over-expression or application of Nur77 agonist cytosporone B suppressed the expression of proinflammatory genes, such as inducible nitric oxide NOS, cyclooxygenase-2, IL-1β, and tumor necrosis factor-α in the activated microglia, while silenced Nur77 exaggerated the inflammatory responses in microglia. Moreover, activation of Nur77 suppressed the LPS-induced NF-κB activation which was partly dependent on p38 MAPK activity, since inhibition of p38 MAPK by SB203580 abolished the LPS-activated NF-κB in microglia. On the other hand, inhibition of p38 MAPK attenuated LPS-induced Nur77 reduction. Furthermore, in a microglia-conditioned cultured media system, Nur77 ameliorated the cytotoxicity to MN9D dopaminergic cells. Lastly, cytosporone B attenuated microglia activation and loss of dopaminergic neuron in the substantia nigra pars compacta (SNpc) of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-PD mouse model. Taken together, these findings revealed the first evidence that Nur77 was an important modulator in microglia function that associated with microglia-mediated dopaminergic neurotoxicity, and thus modulation of Nur77 may represent a potential novel target for treatment for neurodegenerative disease.
Co-reporter:Yafei Zhao, Panpan Wang, Shuangshuang Chen, Chaojun Han, Qiuting Yan, Longtai Zheng, Jia Jia, Zhaoxiang Ren, Xuechu Zhen
Journal of Functional Foods 2017 Volume 33(Volume 33) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.jff.2017.03.034
•Dihydromyricetin (DHM) protected against cerebral ischemia in mouse MCAO model.•Dihydromyricetin (DHM) suppressed microglia-mediated neuroinflammation.•Dihydromyricetin (DHM) activated neuronal ERK-CREB-Bcl-2 signaling pathway.Dihydromyricetin (DHM), a natural flavonoid compound extracted from the fruit Ampelopsis grossedentata, exerts various pharmacological effects. We explored the neuroprotective effects of DHM following cerebral ischemia. Male ICR mice were intraperitoneally injected with DHM for 7 days. Post-ischemic neurological deficits were evaluated with behavioral tests. Mice brain tissues were harvested for infarction analysis, immunohistochemistry. The production of pro-inflammatory mediators in microglial cells were assessed by qPCR and ELISA. Neuroprotective effects of DHM were assessed in HT22 neuronal cells subjected to oxygen-glucose deprivation (OGD)/reoxygenation. DHM reduced the activation of microglia, protected HT22 neurons against OGD-induced injury and promoted functional recovery following middle cerebral artery occlusion (MCAO). Mechanistically, DHM reduced release of pro-inflammatory mediators from microglia. In addition, DHM suppressed caspase-3 cleavage and upregulated CREB, Bcl-2 and phosphorylation of ERK1/2 in HT22 cells. Our study suggested that DHM protects against cerebral ischemia by suppressing microglial neuroinflammation and activating neuronal ERK-CREB-Bcl-2 signaling pathway.
Co-reporter:Zhaohui Yang, Linlang Li, Jiyue Zheng, Haikuo Ma, Sheng Tian, Jiajun Li, Hongjian Zhang, Xuechu Zhen, and Xiaohu Zhang
ACS Chemical Neuroscience 2016 Volume 7(Issue 11) pp:1575
Publication Date(Web):August 28, 2016
DOI:10.1021/acschemneuro.6b00218
Adenosine receptor A2A antagonists have emerged as potential treatment for Parkinson’s disease in the past decade. We have recently reported a series of adenosine receptor antagonists using heterocycles as bioisosteres for a potentially unstable acetamide. These compounds, while showing excellent potency and ligand efficiency, suffered from moderate cytochrome P450 inhibition and high clearance. Here we report a new series of adenosine receptor A2A antagonists based on a 4-amino-5-carbonitrile pyrimidine template. Compounds from this new template exhibit excellent potency and ligand efficiency with low cytochrome P450 inhibition. Although the clearance remains moderate to high, the leading compound, when dosed orally as low as 3 mg/kg, demonstrated excellent efficacy in the haloperidol induced catalepsy rat model for Parkinson’s disease.Keywords: Adenosine receptor; antagonist; GPCR; lead identification; Parkinson’s disease
Co-reporter:Lin Guo;Yanke Chen;Rui Zhao;Guanghui Wang;Eitan Friedman;Ao Zhang
British Journal of Pharmacology 2015 Volume 172( Issue 16) pp:4052-4065
Publication Date(Web):
DOI:10.1111/bph.13195
Background and Purpose
Application of orthosteric sigma-1 receptor agonists as anti-seizure drugs has been hindered by questionable efficacy and potential adverse effects. Here, we have investigated the anti-seizure effects of the novel and potent allosteric modulator of sigma-1 receptors, SKF83959 and its derivative SOMCL-668 (3-methyl-phenyl-2,3,4,5-tetrahydro-1H-benzo[d]azepin-7-ol).
Experimental Approach
The anti-seizure effects of SKF83959 were investigated in three mouse models, maximal electroshock seizures, pentylenetetrazole-induced convulsions and kainic acid-induced ‘status epilepticus’. Also, in rats, the cortical epileptiform activity induced by topical application of picrotoxin was recorded in electrocorticograms. In rat hippocampal brain slices, effects of the drugs on the high potassium-evoked epileptiform local field potentials were studied. Anti-seizure activities of SOMCL-668, a newly developed sigma-1 receptor selective allosteric modulator, were also investigated.
Key Results
SKF83959 (20, 40 mg·kg−1) exhibited anti –seizure actitity in the three mouse models and reduced the cortical epileptiform activity without alteration of spontaneous motor activity and motor coordination. These effects were blocked by the sigma-1 receptor antagonist BD1047, but not the dopamine D1 receptor antagonist SCH23390. SKF83959 alone did not directly inhibit the epileptiform firing of CA3 neurons induced by high potassium in hippocampal slices, but did potentiate inhibition by the orthosteric sigma-1 receptor agonist SKF10047. Lastly, a selective sigma-1 receptor allosteric modulator SOMCL-668, which does not bind to dopamine receptors, exerted similar anti-seizure activities.
Conclusions and Implications
SKF83959 and SOMCL-668 displayed anti-seizure activities, indicating that allosteric modulation of sigma-1 receptors may provide a novel approach for discovering new anti-seizure drugs.
Co-reporter:Peng Lian; LinLang Li; Chuanrong Geng; Xuechu Zhen;Wei Fu
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 8) pp:1616-1627
Publication Date(Web):July 1, 2015
DOI:10.1021/acs.jcim.5b00164
Discovery of high-affinity and high-selectivity agonists of 5-HT1AR has become very attractive due to their potential therapeutic effects on multiple 5-HT1AR-related psychological and neurological problems. On the basis of our previously designed lead compound FW01 (Ki = 51.9 nM, denoted as 9a in the present study), we performed large-scale molecular dynamics simulations and molecular docking operations on 5-HT1AR-9a binding. We found the flip-packing events for the headgroup of 9a, and we also found that its tail group could bind flexibly at the agonist-binding site of 5-HT1AR. By finely tuning the flip-packing phenomenon of the 9a headgroup and tuning the binding flexibility of 9a tail group, we virtually designed a series of new 9a derivatives through molecular docking operations and first-principles calculations and predicted that these newly designed 9a derivatives should be higher-affinity agonists of 5-HT1AR. The computational predictions on the new 9a derivatives have been confirmed by our wet-experimental studies as chemical synthesis, binding affinity assays, and agonistic-function assays. The consistency between our computational design and wet-experimental measurements has led to our discovery of higher-affinity agonists of 5-HT1AR, with ∼50-fold increase in receptor-binding affinity and ∼25-fold improvements in agonistic function. In addition, our newly designed 5-HT1AR agonists showed very high selectivity of 5-HT1AR over subtype 5-HT2AR and also over three subtypes of dopamine receptors (D1, D2, and D3).
Co-reporter:Yu Zhang; Lei Xu; Zhiqiang Zhang; Zhiyu Zhang; Longtai Zheng; Dan Li; Youyong Li; Feng Liu; Kunqian Yu; Tingjun Hou
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 9) pp:1994-2004
Publication Date(Web):August 19, 2015
DOI:10.1021/acs.jcim.5b00445
Macrophage migration inhibitory factor (MIF), a proinflammatory cytokine, is an attractive therapeutic target for the treatment of inflammatory diseases. In our previous study, 3-[(biphenyl-4-ylcarbonyl)carbamothioyl]amino benzoic acid (compound 1) was discovered as a potent inhibitor of MIF by docking-based virtual screening and bioassays. Here, a series of analogues of compound 1 derived from similarity search and chemical synthesis were evaluated for their MIF tautomerase activities, and their structure–activity relationships were then analyzed. The most potent inhibitor (compound 5) with an IC50 of 370 nM strongly suppressed lipopolysaccharide (LPS)-induced production of TNF-α and IL-6 in a dose-dependent manner and significantly enhanced the survival rate of mice with LPS-induced endotoxic shock from 0 to 35% at 0.5 mg/kg and to 45% at 1 mg/kg, highlighting the therapeutic potential of the MIF tautomerase inhibition in inflammatory diseases.
Co-reporter:Zhuang Wu;Linlang Li;Long-Tai Zheng;Zhihong Xu;Lin Guo
Journal of Neurochemistry 2015 Volume 134( Issue 5) pp:904-914
Publication Date(Web):
DOI:10.1111/jnc.13182
Co-reporter:Huali Shi, Sheng Tian, Youyong Li, Dan Li, Huidong Yu, Xuechu Zhen, and Tingjun Hou
Chemical Research in Toxicology 2015 Volume 28(Issue 1) pp:116
Publication Date(Web):December 12, 2014
DOI:10.1021/tx500389q
The activation of pregnane X receptor (PXR), a member of the nuclear receptor (NR) superfamily, can mediate potential drug–drug interactions, and therefore, prediction of PXR activation is of great importance for evaluating drug metabolism and toxicity. In this study, based on 532 structurally diverse compounds, we present a comprehensive analysis with the aim to build accurate classification models for distinguishing PXR activators from nonactivators by using a naive Bayesian classification technique. First, the distributions of eight important molecular physicochemical properties of PXR activators versus nonactivators were compared, illustrating that the hydrophobicity-related molecular descriptors (AlogP and log D) show slightly better capability to discriminate PXR activators from nonactivators than the others. Then, based on molecular physicochemical properties, VolSurf descriptors, and molecular fingerprints, naive Bayesian classifiers were developed to separate PXR activators from nonactivators. The results demonstrate that the introduction of molecular fingerprints is quite essential to enhance the prediction accuracy of the classifiers. The best Bayesian classifier based on the 21 physicochemical properties, VolSurf descriptors, and LCFC_10 fingerprints descriptors yields a prediction accuracy of 92.7% for the training set based on leave-one-out (LOO) cross-validation and of 85.2% for the test set. Moreover, by exploring the important structural fragments derived from the best Bayesian classifier, we observed that flexibility is an important structural pattern for PXR activation. In addition, chemical compounds containing more halogen atoms, unsaturated alkanes chains relevant to π–π stacking, and fewer nitrogen atoms tend to be PXR activators. We believe that the naive Bayesian classifier can be used as a reliable virtual screening tool to predict PXR activation in the drug design and discovery pipeline.
Co-reporter:Xinxian Deng, Lin Guo, Lili Xu, Xuechu Zhen, Kunqian Yu, Weili Zhao, Wei Fu
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 18) pp:3970-3974
Publication Date(Web):15 September 2015
DOI:10.1016/j.bmcl.2015.07.030
A series of compounds with quinazoline scaffold were designed, synthesized and evaluated as novel potent 5-HT2A receptor ligands. N-(4-Chlorophenyl)-2-(piperazin-1-yl)quinazolin-4-amine (5o) has a Ki value of 14.04 ± 0.21 nM, with a selectivity more than 10,000 fold over 5-HT1A receptors (D1 and D2-like receptors). The functional assay showed that this compound is an antagonist to 5-HT2A receptor with an IC50 value of 1.66 μM.
Co-reporter:Lei Xu ; Yu Zhang ; Longtai Zheng ; Chunhua Qiao ; Youyong Li ; Dan Li ; Xuechu Zhen ;Tingjun Hou
Journal of Medicinal Chemistry 2014 Volume 57(Issue 9) pp:3737-3745
Publication Date(Web):April 9, 2014
DOI:10.1021/jm401908w
Macrophage migration inhibitory factor (MIF) is involved in regulation of both the innate and the adaptive immune responses and is regarded as an attractive anti-inflammatory pharmacological target. In this study, molecular docking-based virtual screening and in vitro bioassays were utilized to identify novel small-molecule inhibitors of MIF. The in vitro enzyme-based assay identified that ten chemically diverse compounds exhibited potent inhibitory activity against MIF in the micromolar regime, including three compounds with IC50 values below 10 μM and one with an IC50 value below 1 μM (0.55 μM); the latter is 26-fold more potent than the reference compound ISO-1. The structural analysis demonstrates that most of these active compounds possess novel structural scaffolds. Further in vitro cell-based glucocorticoid overriding, chemotaxis, and Western blotting assays revealed that the three compounds can effectively inhibit the biological functions of MIF in vitro, suggesting that these compounds could be potential agents for treating inflammatory diseases.
Co-reporter:Sheng Tian, Huiyong Sun, Peichen Pan, Dan Li, Xuechu Zhen, Youyong Li, and Tingjun Hou
Journal of Chemical Information and Modeling 2014 Volume 54(Issue 10) pp:2664-2679
Publication Date(Web):September 18, 2014
DOI:10.1021/ci500414b
In this study, to accommodate receptor flexibility, based on multiple receptor conformations, a novel ensemble docking protocol was developed by using the naïve Bayesian classification technique, and it was evaluated in terms of the prediction accuracy of docking-based virtual screening (VS) of three important targets in the kinase family: ALK, CDK2, and VEGFR2. First, for each target, the representative crystal structures were selected by structural clustering, and the capability of molecular docking based on each representative structure to discriminate inhibitors from non-inhibitors was examined. Then, for each target, 50 ns molecular dynamics (MD) simulations were carried out to generate an ensemble of the conformations, and multiple representative structures/snapshots were extracted from each MD trajectory by structural clustering. On average, the representative crystal structures outperform the representative structures extracted from MD simulations in terms of the capabilities to separate inhibitors from non-inhibitors. Finally, by using the naïve Bayesian classification technique, an integrated VS strategy was developed to combine the prediction results of molecular docking based on different representative conformations chosen from crystal structures and MD trajectories. It was encouraging to observe that the integrated VS strategy yields better performance than the docking-based VS based on any single rigid conformation. This novel protocol may provide an improvement over existing strategies to search for more diverse and promising active compounds for a target of interest.
Co-reporter:Jiyue Zheng, Zhaohui Yang, Xuan Li, Linlang Li, Haikuo Ma, Meiyu Wang, Hongjian Zhang, Xuechu Zhen, and Xiaohu Zhang
ACS Chemical Neuroscience 2014 Volume 5(Issue 8) pp:674
Publication Date(Web):June 12, 2014
DOI:10.1021/cn5000716
Parkinson’s disease is a neurodegenerative disease characterized by the motor symptoms of bradykinesia, tremor, and rigidity. Current therapies are based mainly on dopaminergic replacement strategies by administration of either dopamine agonists or dopamine precursor levodopa (L-Dopa). These treatments provide symptomatic relief without slowing or stopping the disease progression, and long-term usage of these drugs is associated with diminished efficacy, motor fluctuation, and dyskinisia. Unfortunately, there had been few novel treatments developed in the past decades. Among nondopaminergic strategies for the treatment of Parkinson’s disease, antagonism of the adenosine A2A receptor has emerged to show great potential. Here we report the optimization of a new chemical scaffold, which achieved exceptional receptor binding affinity and ligand efficiency against adenosine A2A receptor. The leading compounds demonstrated excellent efficacy in the haloperidol induced catalepsy model for Parkinson’s disease.Keywords: Adenosine receptor; antagonist; GPCR; lead optimization; Parkinson’s disease
Co-reporter:Jing Zhang, Jiye Huang, Zilan Song, Lin Guo, Wenxian Cai, Yun Wang, Xuechu Zhen, Ao Zhang
European Journal of Medicinal Chemistry 2014 Volume 85() pp:16-26
Publication Date(Web):6 October 2014
DOI:10.1016/j.ejmech.2014.07.059
•Aryl-N3-benzazepine framework is the prototypical dopamine D1 receptor ligand.•Our study directly focused on modification of the metabolic site-catechol moiety.•Compounds 13b–d displayed Ki values of 270–370 nM at the D1 receptor.•Most compounds possess high affinity less than 10 nM at the 5-HT2A receptor.•13a was the most potent with a Ki value of 4.5 nM at the 5-HT2A receptor.A series of new benzazepines with modification on the catecholic fragment were designed. The 8-hydroxyl group, other than the 7-hydroxyl was confirmed crucial to the interaction with the dopamine D1 receptor. Subsequent replacement of the 7-hydroxyl with benzylamino groups was found tolerable. 7-(m-Chlorophenyl)methylamino- and 7-(m- or o-tolyl)methylamino-substituted benzazepines 13b–d displayed Ki values of 270–370 nM at the D1 receptor, which were slightly more potent than that of parent compound 1. In addition, 7-(arylmethyl)amino-benzazepines 13a–c were found possessing high binding affinities less than 10 nM at the 5-HT2A receptor. Among them, the non-substituted 7-benzylamino analogue 13a was the most potent showing a Ki values of 4.5 nM at the 5-HT2A receptor and a 5-HT2A/D1 selectivity of 147.
Co-reporter:Zeng Li, Jiye Huang, Haifeng Sun, Shengbin Zhou, Lin Guo, Yu Zhou, Xuechu Zhen, Hong Liu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 21) pp:5838-5846
Publication Date(Web):1 November 2014
DOI:10.1016/j.bmc.2014.09.024
A novel scaffold derived from l-SPD with a substituted thiophene group in the D ring were designed, synthesized, and evaluated for their binding affinities at dopamine (D1, D2 and D3) and serotonin (5-HT1A and 5-HT2A) receptors. Most of the tetracyclic compounds exhibited higher affinities for D2 and 5-HT1A receptors than l-SPD, while compound 23e showed the highest Ki value of 7.54 nM at D2 receptor which was 14 times more potent than l-SPD. Additionally, compounds 23d and 23e were more potent than l-SPD at D3 receptor. According to the functional assays, 23d and 23e were demonstrated as full antagonists at D1 and D2 receptors and full agonists at 5-HT1A receptor. Since the combination of D2 antagonism and 5-HT1A agonism is considered effective in treating both the positive and negative symptoms of schizophrenia, these novel compounds are implicated as potential therapeutic agents.
Co-reporter:Ya-Xi Yang, Long-Tai Zheng, Jing-Jing Shi, Bo Gao, Yan-Ke Chen, Hui-Chi Yang, Hong-Li Chen, Yuan-Chao Li, Xue-Chu Zhen
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 4) pp:1222-1227
Publication Date(Web):15 February 2014
DOI:10.1016/j.bmcl.2013.12.055
Glial activation-mediated neuroinflammation plays a pivotal role in the process of several neuroinflammatory diseases including stroke, Alzheimer’s diseases, Parkinson’s diseases, multiple sclerosis and ischemia. Inhibition of microglial activation may ameliorate neuronal degeneration under the inflammatory conditions. In the present study, a number of 5α-cholestan-6-one derivatives were prepared and the anti-inflammatory effects of these compounds were evaluated in LPS-stimulated BV-2 microglia cells. Those derivatives were synthesized from readily available hyodeoxycholic acid (1). Among the tested compounds, several analogs (16–18, 25, 35, 38) exhibited potent inhibitory activities on nitric oxide production with no or weak cell toxicity. Compound 16 also significantly suppressed the expression of TNF-α, interleukin (IL)-1β, cyclooxygenase (COX-2) as well as inducible nitric oxide synthase (iNOS) in LPS-stimulated BV-2 microglia cells. In addition, compound 16 markedly reduced infarction volume in a focal ischemic mice model.
Co-reporter:Lili Xu, Shanglin Zhou, Kunqian Yu, Bo Gao, Hualiang Jiang, Xuechu Zhen, and Wei Fu
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 12) pp:3202-3211
Publication Date(Web):November 18, 2013
DOI:10.1021/ci400481p
The serotonin receptor subtype 1A (5-HT1AR) has been implicated in several neurological conditions, and potent 5-HT1AR agonists have therapeutic potential for the treatment of depression, anxiety, schizophrenia, and Parkinson’s disease. In the present study, a homology model of 5-HT1AR was built based on the latest released high-resolution crystal structure of the β2AR in its active state (PDB: 3SN6). A dynamic pharmacophore model, which takes the receptor flexibility into account, was constructed, validated, and applied to our dynamic pharmacophore-based virtual screening approach with the aim to identify potential 5-HT1AR agonists. The obtained hits were subjected to 5-HT1AR binding and functional assays, and 10 compounds with medium or high Ki and EC50 values were identified. Among them, FW01 (Ki = 51.9 nM, EC50 = 7 nM) was evaluated as the strongest agonist for 5-HT1AR. The active 5-HT1AR model and dynamic pharmacophore model obtained from this study can be used for future discovery and design of novel 5-HT1AR agonists. Also, by integrating all computational and available experimental data, a stepwise 5-HT1AR signal transduction model induced by agonist FW01 was proposed.
Co-reporter:Haifeng Sun, Liyuan Zhu, Huicui Yang, Wangke Qian, Lin Guo, Shengbin Zhou, Bo Gao, Zeng Li, Yu Zhou, Hualiang Jiang, Kaixian Chen, Xuechu Zhen, Hong Liu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 4) pp:856-868
Publication Date(Web):15 February 2013
DOI:10.1016/j.bmc.2012.12.016
An effective and rapid method for the microwave-assisted preparation of the key intermediate for the total synthesis of tetrahydroprotoberberines (THPBs) including l-stepholidine (l-SPD) was developed. Thirty-one THPB derivatives with diverse substituents on A and D ring were synthesized, and their binding affinity to dopamine D1, D2 and serotonin 5-HT1A and 5-HT2A receptors were determined. Compounds 18k and 18m were identified as partial agonists at the D1 receptor with Ki values of 50 and 6.3 nM, while both compounds act as D2 receptor antagonists (Ki = 305 and 145 nM, respectively) and 5-HT1A receptor full agonists (Ki = 149 and 908 nM, respectively). These two THPBs compounds exerted antipsychotic actions in animal models. Further electrophysiological studies employing single-unit recording in intact animals demonstrated that 18k-excited dopaminergic (DA) neurons are associated with its 5-HT1A receptor agonistic activity. These results suggest that these two compounds targeted to multiple neurotransmitter receptors may present novel lead drugs with new pharmacological profiles for the treatment of schizophrenia.
Co-reporter:Wangke Qian, Weijian Lu, Haifeng Sun, Zeng Li, Liyuan Zhu, Rui Zhao, Lei Zhang, Shengbin Zhou, Yu Zhou, Hualiang Jiang, Xuechu Zhen, Hong Liu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 15) pp:4862-4871
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmc.2012.05.057
A series of new tetrahydroprotoberberine (THPB) derivatives were designed, synthesized, and tested for their binding affinity towards dopamine (D1 and D2) and serotonin (5-HT1A and 5-HT2A) receptors. Many of the THPB compounds exhibited high binding affinity and activity at the dopamine D1 receptor, as well as high selectivity for the D1 receptor over the D2, 5-HT1A, and 5-HT2A receptors. Among these, compound 19c exhibited a promising D1 receptor binding affinity (Ki = 2.53 nM) and remarkable selectivity versus D2R (inhibition = 81.87%), 5-HT1AR (inhibition = 61.70%), and 5-HT2AR (inhibition = 24.96%). Compared with l-(S)-stepholidine (l-SPD) (D1Ki = 6.23 nM, D2Ki = 56.17 nM), compound 19c showed better binding affinity for the D1 receptor (2.5-fold higher) and excellent D2/D1 selectivity. Functional assays found compounds 18j, 18k, and 19c are pure D1 receptor antagonists. These results indicate that removing the C10 hydroxy group and introducing a methoxy group at C11 of the pharmacophore of l-SPD can reverse the function of THPB compounds at the D1 receptor. These results are in accord with molecular docking studies.
Co-reporter:Changliang Liu, Xing Fang, Qianqian Wu, Guozhang Jin, Xuechu Zhen
Neuropharmacology (August 2015) Volume 95() pp:299-308
Publication Date(Web):1 August 2015
DOI:10.1016/j.neuropharm.2015.03.037
•Prefrontal cortex (PFC) is important for morphine to excite dopamine neurons.•PFC affects morphine-induced dopamine release.•Morphine exposure changes the role of PFC in morphine action.•Morphine exposure alters the response of PFC pyramidal neurons to morphine.Morphine excites dopamine (DA) neurons in the ventral tegmental area (VTA), an effect mediated by both local and systemic mechanisms. While the importance of the prefrontal cortex (PFC) – VTA circuit in opiate addiction is well established, little is known about how the PFC regulates the activity of VTA DA neurons upon morphine stimulation. One major challenge is that VTA DA neurons are highly heterogeneous in terms of projection and regulation, making their responses to PFC manipulations variable. Our previous work has identified a subgroup of VTA DA neurons exhibiting significant slow oscillation in their firing sequence, and demonstrated that most of these neurons are functionally connected with the PFC. In the present study, we focus our efforts only on VTA DA neurons expressing strong slow oscillation, and report that blocking the neuronal activity in the PFC remarkably attenuates the morphine-induced excitation of these neurons. Using in vivo microdialysis, we find that inactivation of the PFC also reduces the morphine-induced elevation of DA levels in the nucleus accumbens (NAc). Furthermore, 24 h after only single morphine exposure, PFC-inactivation failed to prevent subsequent morphine challenge from exciting VTA DA neurons, which is paralleled by altered response of PFC pyramidal neurons to morphine stimulation. Our results indicate that the PFC gates acute morphine action on a subset of VTA DA neurons, which is highly plastic and can be functionally remodeled by morphine exposure.
Co-reporter:Qin Jin, Jian Cheng, Yang Liu, Jian Wu, Xiaoyu Wang, Shanwen Wei, Xiaomei Zhou, Zhenghong Qin, Jia Jia, Xuechu Zhen
Brain, Behavior, and Immunity (August 2014) Volume 40() pp:131-142
Publication Date(Web):August 2014
DOI:10.1016/j.bbi.2014.03.003

![1H-Indole-6-carboxylic acid, 2,3-dihydro-3-[[[4-[methyl[2-(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-, methyl ester](http://img.cochemist.com/ccimg/1160300/1160294-26-7.png)
![1H-Indole-6-carboxylic acid, 2,3-dihydro-3-[[[4-[methyl[2-(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-, methyl ester](http://img.cochemist.com/ccimg/1160300/1160294-26-7_b.png)


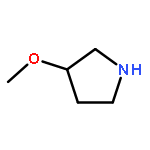





![6-CHLORO-1-CYCLOBUTYL-3-METHYL-1H-PYRROLO[2,3-B]PYRIDINE](http://img.cochemist.com/ccimg/1032100/1032047-47-4.png)
![6-CHLORO-1-CYCLOBUTYL-3-METHYL-1H-PYRROLO[2,3-B]PYRIDINE](http://img.cochemist.com/ccimg/1032100/1032047-47-4_b.png)
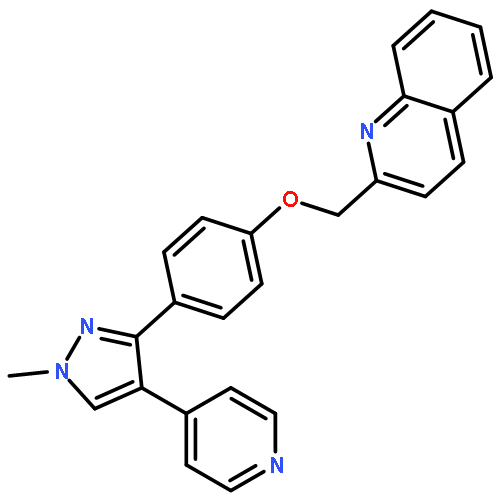


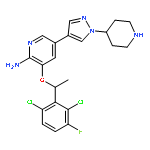
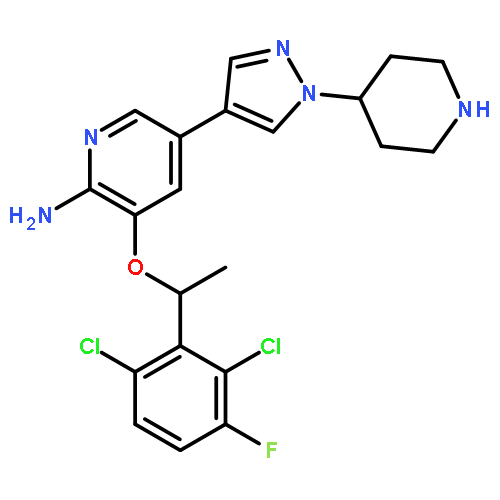




![3-({[(3-chlorobenzoyl)amino]carbonothioyl}amino)benzoic acid](http://img.cochemist.com/ccimg/434000/433942-71-3.png)
![3-({[(3-chlorobenzoyl)amino]carbonothioyl}amino)benzoic acid](http://img.cochemist.com/ccimg/434000/433942-71-3_b.png)
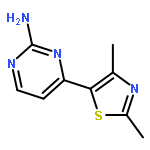
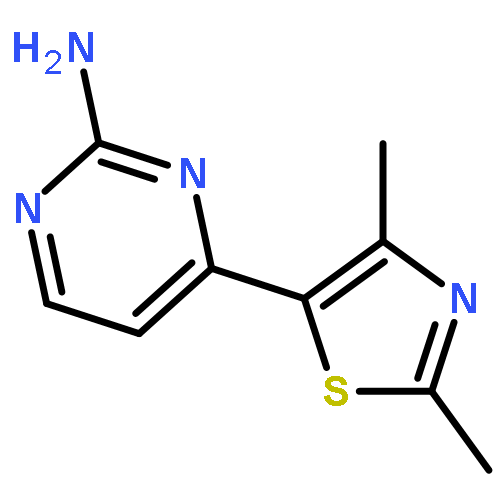




![3',5'-Difluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/350700/350682-84-7.png)
![3',5'-Difluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/350700/350682-84-7_b.png)












![3',5'-Dichloro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/191000/190911-79-6.png)
![3',5'-Dichloro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/191000/190911-79-6_b.png)
![2-[[9-(1-Methylethyl)-6-[(phenylmethyl)amino]-9H-purin-2-yl]amino]-1-butanol](http://img.cochemist.com/ccimg/186700/186692-44-4.png)
![2-[[9-(1-Methylethyl)-6-[(phenylmethyl)amino]-9H-purin-2-yl]amino]-1-butanol](http://img.cochemist.com/ccimg/186700/186692-44-4_b.png)


![4-[2-[(6-ethoxy-1H-benzimidazol-2-yl)sulfanyl]ethyl]morpholine](http://img.cochemist.com/ccimg/173400/173352-21-1.png)
![4-[2-[(6-ethoxy-1H-benzimidazol-2-yl)sulfanyl]ethyl]morpholine](http://img.cochemist.com/ccimg/173400/173352-21-1_b.png)




![N-[2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl]-N-pyridin-2-ylcyclohexanecarboxamide](http://img.cochemist.com/ccimg/162800/162760-96-5.png)
![N-[2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl]-N-pyridin-2-ylcyclohexanecarboxamide](http://img.cochemist.com/ccimg/162800/162760-96-5_b.png)






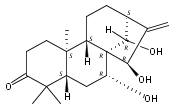












![Benzoic acid,4-[[(benzoylamino)thioxomethyl]amino]-, ethyl ester](http://img.cochemist.com/ccimg/69200/69165-46-4.png)
![Benzoic acid,4-[[(benzoylamino)thioxomethyl]amino]-, ethyl ester](http://img.cochemist.com/ccimg/69200/69165-46-4_b.png)


![Sto-609 Acetate;7-oxo-7h-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylicacidacetate](http://img.cochemist.com/ccimg/52100/52029-86-4.png)
![Sto-609 Acetate;7-oxo-7h-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylicacidacetate](http://img.cochemist.com/ccimg/52100/52029-86-4_b.png)
![Benzoic acid, 4-[[(benzoylamino)thioxomethyl]amino]-](http://img.cochemist.com/ccimg/40700/40611-30-1.png)
![Benzoic acid, 4-[[(benzoylamino)thioxomethyl]amino]-](http://img.cochemist.com/ccimg/40700/40611-30-1_b.png)
















![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)