Co-reporter:Xufeng Cao ; Zhaoshuan Sun ; Yongbing Cao ; Ruilian Wang ; Tongkai Cai ; Wenjing Chu ; Wenhao Hu
Journal of Medicinal Chemistry 2014 Volume 57(Issue 9) pp:3687-3706
Publication Date(Web):February 24, 2014
DOI:10.1021/jm4016284
Triazoles with fused-heterocycle nuclei were designed and evaluated for their in vitro activity on the basis of the binding mode of albaconazole using molecular docking, along with SAR of antifungal triazoles. Tetrahydro-[1,2,4]triazolo[1,5-a]pyrazine and tetrahydro-thiazolo[5,4-c]pyridine nuclei were preferable to the other four fused-heterocycle nuclei investigated. Potent in vitro activity, broad spectrum and better water solubility were attained when triazoles containing nitrogen aromatic heterocycles were attached to these two nuclei. The most potent compounds 27aa and 45x, with low hERG inhibition and hepatocyte toxicity, both exhibited excellent activity against Candida, Cryptococcus, and Aspergillus spp., as well as selected fluconazole-resistant strains. A high water-soluble compound 58 (the disulfate salt of 45x) displayed unsatisfactory in vivo activity because of its poor PK profiles. Mice infected with C.alb. SC5314 and C.alb. 103 (fluconazole-resistant strain) and administered with 27aa displayed significantly improved survival rates. 27aa also showed favorable pharmacokinetic (PK) profiles.
Co-reporter:Tao Xue ; Shi Ding ; Bin Guo ; Yuren Zhou ; Peng Sun ; Heyao Wang ; Wenjing Chu ; Guoqing Gong ; Yinye Wang ; Xiaoyan Chen
Journal of Medicinal Chemistry 2014 Volume 57(Issue 18) pp:7770-7791
Publication Date(Web):September 2, 2014
DOI:10.1021/jm501045e
The blood coagulation enzyme factor Xa (FXa) is a particularly promising target for anticoagulant therapy, and identification of oral small-molecule inhibitors of FXa remains a research focus. On the basis of the X-ray crystal structure of FXa and its inhibitor rivaroxaban, we designed and synthesized a series of conformationally restricted mimics containing a novel [6,6,5] tricyclic fused oxazolidinone scaffold. Intensive structure–activity relationship (SAR) and structure–pharmacokinetic relationship (SPR) studies on this new series led to the discovery of compound 11a: a highly potent, selective, direct, and orally bioavailable FXa inhibitor with excellent in vivo antithrombotic efficacy and preferable pharmacokinetic profiles. Druggability evaluation of compound 11a was undertaken and elicited positive outcomes. All results indicate that compound 11a is a promising drug candidate for the prevention and treatment of thromboembolic diseases in venous and arterial systems.
Co-reporter:Shouning Yang, Wei Shi, Dong Xing, Zheng Zhao, Fengping Lv, Liping Yang, Yushe Yang, Wenhao Hu
European Journal of Medicinal Chemistry 2014 Volume 86() pp:133-152
Publication Date(Web):30 October 2014
DOI:10.1016/j.ejmech.2014.07.106
•43 formyl hydroxyamino derivatives were rationally designed and synthesized as PDF inhibitors.•The SAR on P1′, P2′ and P3′ positions of the PDF inhibitors were studied.•Full set of biological evaluation were carried out to identify PDF inhibitors with good in-vivo efficacy and low toxicity.Peptide deformylase (PDF) has been identified as a promising target for novel antibacterial agents. In this study, a series of novel formyl hydroxyamino derivatives were designed and synthesized as PDF inhibitors and their antibacterial activities were evaluated. Among the potent PDF inhibitors (1o, 1q, 1o′, 1q′, and 1x), in vivo studies showed that compound 1q possesses mild toxicity, a good pharmacokinetic profile and protective effects. The good in vivo efficacy and low toxicity suggest that this class of compounds has potential for development and use in future antibacterial drugs.
Co-reporter:Suo Gao, Jian-Jun Cheng, Chen-Yu Ling, Wen-Jing Chu, Yu-She Yang
Tetrahedron Letters 2014 Volume 55(Issue 35) pp:4856-4859
Publication Date(Web):27 August 2014
DOI:10.1016/j.tetlet.2014.05.034
A convergent enantioselective total synthesis of (−)-(S)-stepholidine, a drug candidate for the treatment of schizophrenia and/or drug abuse, was described, which represented the first example of successful auxiliary-assisted Bischler–Napieralski cyclization of amide bearing bromine atom at 2-position of the C ring, followed by an introduction of the aryl methyl ester via Br–Li exchange. (−)-(S)-Stepholidine was synthesized in 6 steps, with 52% overall yield and >99% ee. The reported synthesis is practically free from chromatographic separation.
Co-reporter:Bin Guo ; Houxing Fan ; Qisheng Xin ; Wenjing Chu ; Hui Wang ; Yanqin Huang ; Xiaoyan Chen
Journal of Medicinal Chemistry 2013 Volume 56(Issue 6) pp:2642-2650
Publication Date(Web):February 21, 2013
DOI:10.1021/jm4000598
The solubility-driven structural modification of (pyridin-3-yl) benzoxazinyl-oxazolidinones is described, which resulted in the development of a new series of benzoxazinyl-oxazolidinone analogues with high antibacterial activity against Gram-positive pathogens, including that against linezolid-resistant strains and low hERG inhibition. With regard to structure–activity relationship (SAR) trends among the various substituents on the pyridyl ring, relatively small and nonbasic substituents were preferable to sterically demanding or basic substituents. Oxazolidinone ring substitution on the pyridyl ring generated analogues with antibacterial activity superior to imidazolidinone ring. Solubility was enhanced by the incorporation of polar groups, especially when compounds were converted to their prodrugs. Among the prodrugs, compound 85 exhibited excellent solubility and a good pharmacokinetic profile. In a MRSA systemic infection model, compound 85 displayed an ED50 = 5.00 mg/kg, a potency that is 2-fold better than that of linezolid.
Co-reporter:Xu-Feng Cao, Wen-Jing Chu, Yong-Bing Cao, Yu-She Yang
Chinese Chemical Letters 2013 Volume 24(Issue 4) pp:303-306
Publication Date(Web):April 2013
DOI:10.1016/j.cclet.2013.01.047
In order to find novel antifungal agents with good activity and aqueous solubility, a series of SYN-2869 analogues containing a pyridine ring were synthesized and evaluated for their in vitro antifungal activity and water solubility. The results indicated that some compounds showed potent activity against six pathogenic fungi. In particular, the analogue 17a having an isobutyl substitution on the triazolone exhibited significant broad spectrum antifungal activity. In addition, the water solubility of compound 17a was sufficiently improved over SYN-2869.In order to find novel antifungal agents with good activity and aqueous solubility, a series of SYN-2869 analogues containing a pyridine ring were synthesized and evaluated for their in vitro antifungal activity and water solubility. The most potent compound 17a having an isobutyl substitution on the triazolone exhibited significant broad spectrum antifungal activity. In addition, the water solubility of compound 17a was sufficiently improved over SYN-2869.
Co-reporter:Yu Liu, Xu-Feng Cao, Xin Liu, Yong-Bing Cao, Wen-Jing Chu, Yu-She Yang
Chinese Chemical Letters 2013 Volume 24(Issue 4) pp:321-324
Publication Date(Web):April 2013
DOI:10.1016/j.cclet.2013.03.008
To improve the aqueous solubility of an itraconazole analogue, compound 1 (YL-24), a series of novel prodrugs were synthesized. Among these prodrugs, the phosphate disodium salt compound 7 exhibited excellent aqueous solubility (9.8 mg/mL) at near-neutral pH and sufficient stability in buffer solutions, along with favorable pharmacokinetic profiles. In particular, compounds 1 and 7 provided moderate survival efficacy in murine systemic Candida albicans SC5314 infection model, but their efficacy was weaker than that of fluconazole.To improve the aqueous solubility of an itraconazole analogue, compound 1 (YL-24), a series of novel prodrugs were synthesized. Among these prodrugs, the phosphate disodium salt compound 7 exhibited excellent aqueous solubility (9.8 mg/mL) at near-neutral pH and sufficient stability in buffer solutions, along with favorable pharmacokinetic profiles. In particular, compounds 1 and 7 provided moderate survival efficacy in murine systemic Candida albicans SC5314 infection model, but their efficacy was weaker than that of fluconazole.
Co-reporter:XiaoDan Fu, XingQun Guo, XingWei Li, LiDong He, YuShe Yang, YouXi Chen
Tetrahedron: Asymmetry 2013 Volume 24(13–14) pp:827-832
Publication Date(Web):31 July 2013
DOI:10.1016/j.tetasy.2013.05.006
An asymmetric synthesis of the melatonin receptor agonist Ramelteon 1 has been achieved, which involved a tandem C–H activation–alkylation/Heck reaction and subsequent highly diastereoselective asymmetric Michael addition to generate the corresponding chiral intermediate, which was readily converted into Ramelteon 1 in 19% overall yield in 15 linear steps.(E)-Ethyl 3-(5-(1-(S)-((tert-butylsulfinyl)imino)ethyl)-2,3-dihydrobenzofuran-4-yl) acrylateC19H25NO4S[α]D25=-4.1 (c 1.0, CHCl3)Absolute configuration: (E,S)Ethyl 2-((8S)-6-(((S)-tert-butylsulfinyl)imino)-2,6,7,8-tetrahydro-1H-indeno[5,4-b]furan-8-yl)acetateC19H25NO4S[α]D25=+96.9 (c 1.0, CHCl3)Absolute configuration: (8S,S)(S)-Ethyl 2-(6-oxo-2,6,7,8-tetrahydro-1H-indeno[5,4-b]furan-8-yl)acetate.C15H16O4[α]D25=-73.5 (c 1.0, CHCl3)ee value: 92%Absolute configuration: (S)(S)-Ethyl 2-(2,6,7,8-tetrahydro-1H-indeno[5,4-b]furan-8-yl)acetate.C15H18O3[α]D25=-58.9 (c 1.0, CHCl3)Absolute configuration: (S)(S)-2-(2,6,7,8-Tetrahydro-1H-indeno[5,4-b]furan-8-yl)ethanol.C13H16O2[α]D25=-81.4 (c 1.0, CHCl3)Absolute configuration: (S)(S)-2-(2,6,7,8-Tetrahydro-1H-indeno[5,4-b]furan-8-yl)ethanamine.C13H17NO3[α]D25=-69.2 (c 1.0, CHCl3)Absolute configuration: (S)RamelteonC16H21NO2[α]D25=-54.8 (c 1.0, CHCl3)
Co-reporter:Liqiang Fu, Xin Liu, Chenyu Ling, Jianjun Cheng, Xingsheng Guo, Huili He, Shi Ding, Yushe Yang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 2) pp:814-819
Publication Date(Web):15 January 2012
DOI:10.1016/j.bmcl.2011.12.063
We report herein the design, synthesis, and structure–activity relationship studies of conformationally restricted mutilin 14-carbamates based on the structure of SB-222734. The antibacterial activities of these newly synthesized compounds were also evaluated and compared with linezolid and retapamulin. Results showed that most of the target compounds exhibit good potency in inhibiting the growth of Gram-positive bacteria including Methicillin-susceptible Staphylococcus aureus MSSA (MIC: 0.0625–2 μg/mL), Methicillin-resistant S. aureus MRSA (MIC: 0.0625–2 μg/mL), Methicillin-susceptible Staphylococcus epidermidis MSSE (MIC: 0.0625–2 μg/mL), Methicillin-resistant S. epidermidis MRSE (MIC: 0.0625–2 μg/mL), and Streptococcus pneumonia (MIC: 0.0625–4 μg/mL). In particular, three remarkable compounds of this series (12l, 12m, and 21l) exhibited comparable in vitro antibacterial profiles to that of retapamulin.
Co-reporter:Li Qiang Fu, Chen Yu Ling, Xing Sheng Guo, Hui Li He, Yu She Yang
Chinese Chemical Letters 2012 Volume 23(Issue 1) pp:9-12
Publication Date(Web):January 2012
DOI:10.1016/j.cclet.2011.10.002
In order to find novel antibacterial agents with superior antibacterial activity and overcoming multidrug resistance, a series of pleuromutilin derivatives with novel C(14) side chain were synthesized and evaluated for their in vitro antibacterial activities. The results of antibacterial acticities indicated that most of the derivatives showed potent activities against Gram-positive organisms. In particular, compound 10d exhibited the most potent inhibitory activity compared with pleuromutilin and linezoid, emerged as potential molecule for further investigation.
Co-reporter:Qisheng Xin ; Houxing Fan ; Bin Guo ; Huili He ; Suo Gao ; Hui Wang ; Yanqin Huang
Journal of Medicinal Chemistry 2011 Volume 54(Issue 21) pp:7493-7502
Publication Date(Web):September 28, 2011
DOI:10.1021/jm200614t
A series of novel benzoxazinyl-oxazolidinones bearing nonaromatic heterocycle or aryl groups were designed and synthesized. Their in vitro and in vivo antibacterial activities were investigated. Most of the (3S, 3aS) biaryl benzoxazinyl-oxazolidinones exhibited potent activity against Gram-positive pathogens. SAR trends were observed; a pyridyl C ring was preferable to other 5- or 6-member aryl C rings, while fluorine substitution on the B ring generated derivatives with reduced activity. Various substituent group positions on the pyridyl ring were also evaluated. The resulting compounds displayed excellent activity against linezolid-resistant strains. Compound 45 exhibited excellent in vitro activity, with a MIC value of 0.25–0.5 μg/mL against MRSA and an activity against linezolid-resistant strains of 8–16-fold higher potency than linezolid. In a MRSA systemic infection model, compound 45 displayed an ED50 < 5.0 mg/kg, a potency that is nearly 3-fold better than that of linezolid. This compound also showed excellent pharmacokinetic profiles, with a half-life of more than 5 h as well as an oral bioavailability of 81% in rats.
Co-reporter:Yu Liu, Zining Liu, Xufeng Cao, Xin Liu, Huili He, Yushe Yang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 16) pp:4779-4783
Publication Date(Web):15 August 2011
DOI:10.1016/j.bmcl.2011.06.062
To improve antifungal activities, water solubility and bioavailability, a series of novel analogues of itraconazole-containing pyridine rings were designed and synthesized. Their antifungal activities were evaluated in vitro against six clinically important fungi by measuring the minimal inhibitory concentrations (MICs). Most of the compounds showed more potent antifungal activities than that of itraconazole. In particular, the analogues 30d, 30c, 31c, and 36d exhibited much higher solubility and bioavailability than that of itraconazole. The bioavailability of 36d (42.2%) was five times higher than that of itraconazole (8%) and was negative for genetic toxicology in the Ames test.A series of novel analogues of itraconazole-containing pyridine rings were designed and synthesized. Most of the compounds showed more potent antifungal activities than that of the parent itraconazole. In particular, the analogues 30d, 30c, 31c, and 36d exhibited much higher solubility and bioavailability than did itraconazole. The bioavailability of 36d (42.2%) was five times higher than that of itraconazole (8%) and was negative for genetic toxicology in the Ames test.
Co-reporter:Xiao Zhong Fu, Yu Ou, Jan Xin, Yu She Yang
Chinese Chemical Letters 2011 Volume 22(Issue 12) pp:1387-1390
Publication Date(Web):December 2011
DOI:10.1016/j.cclet.2011.09.005
A series of novel mono (2, 2, 2-trifluoroethyl) esters, mono l-amino acid ester prodrugs of acyclic nucleoside phosphonates was synthesized and their in vitro anti-HBVactivity was evaluated in HepG 2 2.2.15 cells. Compound 1d exhibited more potent anti-HBV activity and lower cytotoxicity than those of adefovir dipivoxil and alamifovir (MCC-478) with EC50 and CC50 values of 0.01 μmol/L and >8000 μmol/L respectively.
Co-reporter:Liqiang Fu, Xin Liu, Jianjun Cheng, Huili He, Zhan Cai, Xingsheng Guo, Shi Ding and Yushe Yang
Organic Process Research & Development 2010 Volume 14(Issue 4) pp:815-819
Publication Date(Web):May 12, 2010
DOI:10.1021/op100063h
A first scale-up synthesis of FU-23 (1), a potent and effective water-soluble pleuromutilin-derived antibiotic, is described. The original synthesis from the medicinal chemistry group provided 1 in seven steps and 10.9% overall yield, required four chromatographies and employed expensive reagents such as AgOCN and 4-acetylsalicoyl chloride. The optimized synthetic route for the preparation of phosphate salt 1 consists of seven linear steps with a 42.8% overall yield. Significant cost reduction and more robust reaction conditions have been developed with no chromatography required at any stage.
Co-reporter:Li Qiang Fu, Xin Sheng Guo, Xin Liu, Hui Li He, Yu Lin Wang, Yu She Yang
Chinese Chemical Letters 2010 Volume 21(Issue 5) pp:507-510
Publication Date(Web):May 2010
DOI:10.1016/j.cclet.2010.01.001
In order to probe the effect of C-2(S)-substituted groups in the antibacterial activity, a series of novel C-2(S)-substituted pleuromutilin analogues of SB-225586 were synthesized and evaluated for their in vitro antibacterial activity. The results of antibacterial activities indicated that C-2(S)-substituted pleuromutilin derivatives retained appreciable antibacterial activity, and the 2-fluorination compounds 6a and 6b are more potent than the corresponding 2-hydroxylation analogues 7a and 7b.
Co-reporter:Peng Lu, Sai Hong Jiang, Jiang Xia Liu, Yu She Yang, Ru Yun Ji
Chinese Chemical Letters 2009 Volume 20(Issue 5) pp:507-510
Publication Date(Web):May 2009
DOI:10.1016/j.cclet.2008.12.057
A series of novel bis(trifluoroethyl)phosphonomethyl ether derivatives of acyclovir was synthesized and their in vitro anti-HBV activity was evaluated in HepG2 2.2.15 cells. In contrast to acyclovir, most of the described phosphonates emerged as potent inhibitors of HBV replication. Especially, the most active compound 11 with IC50 value of 2.92 μmol/L was 33 times more potent than acyclovir with IC50 value of 100 μmol/L.
Co-reporter:Liqiang Fu, Zhiteng Jiang, Zhan cai, Xin Liu, Huili He, Yushe Yang
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 18) pp:5407-5410
Publication Date(Web):15 September 2009
DOI:10.1016/j.bmcl.2009.07.115
A phosphate prodrug strategy was investigated to address the problem of poor aqueous solubility of pleuromutilin analogues. Water-soluble phosphate prodrugs 6a, 6b and 6c of pleuromutilin analogues were designed and synthesized. Three compounds all exhibited excellent aqueous solubility (>50 mg/mL) at near-neutral pH and sufficient stability in buffer solution. In particular, the phenol pleuromutilin prodrug 6c displayed favourable pharmacokinetic profiles and comparable potency with vancomycin against MSSA and MRSA strains in vivo.Synthesis and biological properties of phosphate prodrugs of pleuromutilin analogues are disclosed. Compound 6c was metabolized efficiently to the biologically active parent 5d in vivo, it also showed excellent antibacterial activity against MSSA and MRSA with comparable ED50 as vancomycin by iv administration in mice.
Co-reporter:Peng Lu, Jiangxia Liu, Yuya Wang, Xiaoyan Chen, Yushe Yang, Ruyun Ji
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 24) pp:6918-6921
Publication Date(Web):15 December 2009
DOI:10.1016/j.bmcl.2009.10.072
A series of novel oxazaphosphorine prodrugs of 9-(2-phosphonomethoxyethyl)adenine (PMEA, adefovir) were synthesized and their anti-hepatitis B virus (HBV) activity was evaluated in HepG2 2.2.15 cells, with adefovir dipivoxil as a reference drug. In the cell assays, compounds 7b and 7d exhibited anti-HBV activity comparable to that of adefovir dipivoxil, while compound 7c, with an IC50 value of 0.12 μM, was found to be three times more potent than the reference compound. In vitro stability studies showed that (SP,S)-7c, the diastereomer of compound 7c, was stable in human blood plasma but underwent rapid metabolism to release the parent drug PMEA in liver microsomes. The possible metabolic pathway of (SP,S)-7c in human liver microsomes was described. These findings suggest that compound (SP,S)-7c is a promising anti-HBV drug candidate for further development.A series of novel oxazaphosphorine prodrugs of PMEA is disclosed. l-valine methyl ester (7c) demonstrated highly potent anti-HBV activity, excellent stability in human plasma and release of the parent compound PMEA in human microsomes.
Co-reporter:Houxing Fan, Yilang Chen, Zhiteng Jiang, Shuhua Zhang, Dafang Zhong, Ruyun Ji, Yushe Yang
European Journal of Medicinal Chemistry 2008 Volume 43(Issue 8) pp:1706-1714
Publication Date(Web):August 2008
DOI:10.1016/j.ejmech.2007.09.010
A new series of oxazolidinones bearing N-linked 5-triazolylmethyl group have been synthesized and their in vitro antibacterial activities (MIC) were evaluated against a spectrum of resistant and susceptible Gram-positive organisms. Some of the analogues in this series displayed activity superior to linezolid and vancomycin. Furthermore, in vivo efficacies and pharmacokinetic properties of the selected compounds were also disclosed herein; the selected compounds showed reasonable bioavailability as well as in vivo efficacy comparable to that of linezolid.A new series of oxazolidinones bearing N-linked 5-triazolylmethyl group are disclosed. The selected compounds of this series display in vitro and in vivo activities comparable to linezolid. Compound 4e shows excellent activity against Gram-positive organisms.
Co-reporter:Lei Tang, Juanhong Yu, Haoshu Wu, Houxing Fan, Yushe Yang, Ruyun Ji
European Journal of Medicinal Chemistry 2008 Volume 43(Issue 9) pp:1997-2003
Publication Date(Web):September 2008
DOI:10.1016/j.ejmech.2007.11.018
A series of phenylalanine derivatives were synthesized and their biological activities were evaluated. Compounds (S)-3 and (R)-3 exhibited more potent insulin-releasing activity than that of nateglinide, compound (S)-3 also showed insulin-sensitizing activity in vitro. Both compounds were tested for hypoglycemia effect in vivo.
Co-reporter:Juanhong Yu, Lei Tang, Yushe Yang, Ruyun Ji
European Journal of Medicinal Chemistry 2008 Volume 43(Issue 11) pp:2428-2435
Publication Date(Web):November 2008
DOI:10.1016/j.ejmech.2008.01.029
A series of benzopyran derivatives were synthesized and evaluated for PPAR α/γ agonist activities. Most of the compounds exhibit reasonable PPAR α and PPAR γ agonist activities. In particular, compounds 7b, 8b, 8e and 8h with remarkable PPARg EC50 values of 0.001 μM are excellent full PPAR γ agonists with the functional potency about 130, 20 times stronger than that of leading compound 5 and rosiglitazone, respectively. Compounds 7a, 7c, 7d and 8a are dual PPAR α/γ agonists, and all of them gave comparable or stronger PPAR α/γ agonist efficacy than that of the corresponding positive control.A new series of benzopyran derivatives were synthesized as continuing work of compound 5. Their PPAR agonist activities were evaluated and most of the compounds exhibit reasonable PPAR α and PPAR γ agonist activities. In particular, compounds 7b, 8b, 8e and 8h with remarkable PPAR γ EC50 values of 0.001 μM are excellent full PPAR γ agonists.
Co-reporter:Houxing Fan, Gang Xu, Yilang Chen, Zhiteng Jiang, Shuhua Zhang, Yushe Yang, Ruyun Ji
European Journal of Medicinal Chemistry 2007 Volume 42(Issue 8) pp:1137-1143
Publication Date(Web):August 2007
DOI:10.1016/j.ejmech.2007.01.012
A new series of oxazolidinones containing triazolyl group has been synthesized and tested for in vitro antibacterial activity by MIC determination against a panel of resistant and susceptible Gram-positive organisms. Most of the analogs in this series displayed activity superior to linezolid and vancomycin in vitro. Further, in vivo efficacies of the selected oxazolidinones were also disclosed herein.A new series of oxazolidinones containing triazolyl group has been synthesized and their in vitro antibacterial activity (MIC) was evaluated, furthermore, in vivo efficacies of the selected compounds were also disclosed herein.
Co-reporter:Xiao Zhong Fu, Sai Hong Jiang, Jian Xin, Yu She Yang, Ru Yun Ji
Chinese Chemical Letters 2007 Volume 18(Issue 7) pp:817-819
Publication Date(Web):July 2007
DOI:10.1016/j.cclet.2007.05.013
A series of novel l-amino acid esters prodrugs of acyclic nucleoside phosphonates was synthesized and their anti-HBV activity was evaluated in HepG2 2.2.15 cells. Compound 1d exhibited more potent anti-HBV activity and lower cytotoxicity than those of adefovir dipivoxil with EC50 and CC50 values of 0.207 μmol/L and 2530 μmol/L, respectively.
Co-reporter:Xiaozhong Fu, Saihong Jiang, Chuan Li, Jian Xin, Yushe Yang, Ruyun Ji
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 2) pp:465-470
Publication Date(Web):15 January 2007
DOI:10.1016/j.bmcl.2006.10.021
A series of novel bis(l-amino acid) ester prodrugs of 9-[2-(phosphonomethoxy)ethyl] adenine (PMEA) was synthesized and their anti-HBV activity was evaluated in HepG 2 2.2.15 cells. Compounds 11, 12, 21, 22, 26, and 27 demonstrated more potent anti-HBV activity and higher selective index (SI) than adefovir dipivoxil, which was used as a positive control. Compound 11, which was found to be the most potent one, was five times more potent than adefovir dipivoxil with EC50 value of 0.095 μM and CC50 value of 6636 μM. The SI value (>69,000) of compound 11 was 60 times and 24 times higher than those of adefovir dipivoxil and lamivudine, respectively. In vitro stability studies showed that compound 11 was relatively more stable than adefovir dipivoxil with t1/2 of 270 min. These findings suggested that compound 11 could be considered as a promising candidate for further in vivo studies.A series of novel bis(l-amino acid)ester prodrugs of 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA) was synthesized and their anti-HBV activity was evaluated in HepG2 2.2.15 cells. Compound 11 exhibited five times more potent anti-HBV activity, 60 times higher selective index (SI) than adefovir dipivoxil. Moreover, compound 11 was more stable than adefovir dipivoxil with t1/2 of 270 min.
Co-reporter:Zhao Dang, Yushe Yang, Ruyun Ji, Shuhua Zhang
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 16) pp:4523-4526
Publication Date(Web):15 August 2007
DOI:10.1016/j.bmcl.2007.05.093
The design and synthesis of new fluoroquinolone antibacterial agents having substituted piperidine rings at the C-7 position are described. Most of the new compounds demonstrated high in vitro antibacterial activity. Several of them exhibited significant activities against Gram-positive organisms, which were more potent than those of gemifloxacin, Linezolid, and vancomycin.The design, synthesis, and antibacterial activity of new fluoroquinolones possessing substituted piperidine rings at the C-7 position are described.
Co-reporter:Yingjie Cui, Yaxian Dang, Yushe Yang, Shuhua Zhang, Ruyun Ji
European Journal of Medicinal Chemistry 2005 Volume 40(Issue 2) pp:209-214
Publication Date(Web):February 2005
DOI:10.1016/j.ejmech.2004.10.012
The syntheses of substituted piperazinyl pyridyl oxazolidinones 8–16 are described. Their in vitro activities against Gram-positive organisms such as Staphylococcus aureus, Staphylococcus epidermidis, Streptococcus and enterococcus were evaluated by minimum inhibitory concentration (MIC) determination. Compound 8 and 10 were found to be superior to linezolid.
Co-reporter:Tang Lei;Leng Ying;Wang Huo-Quan;Feng Ying;Yang Yu-She;Ji Ru-Yun
Chinese Journal of Chemistry 2003 Volume 21(Issue 4) pp:
Publication Date(Web):26 AUG 2010
DOI:10.1002/cjoc.20030210402
A series of 2-benzyl-1,3-dicarbonyl derivatives was synthesized. Their insulin-sensitizing activity was evaluated in 3T3-L1 preadipocyte cells. Compounds 3, 26 and 27 were found to possess strong insulin-sensitizing activity in vitro and were selected for further hypoglycemic evaluation in vivo.

![(2S)-2-[(tert-Butoxycarbonyl)amino]-3-hydroxypropanoic acid](http://img.cochemist.com/ccimg/1251100/1251002-72-8.png)
![(2S)-2-[(tert-Butoxycarbonyl)amino]-3-hydroxypropanoic acid](http://img.cochemist.com/ccimg/1251100/1251002-72-8_b.png)
![tert-Butyl 6,7-dihydro-[1,2,3]triazolo[1,5-a]pyrazine-5(4H)-carboxylate](http://img.cochemist.com/ccimg/1245800/1245782-69-7.png)
![tert-Butyl 6,7-dihydro-[1,2,3]triazolo[1,5-a]pyrazine-5(4H)-carboxylate](http://img.cochemist.com/ccimg/1245800/1245782-69-7_b.png)
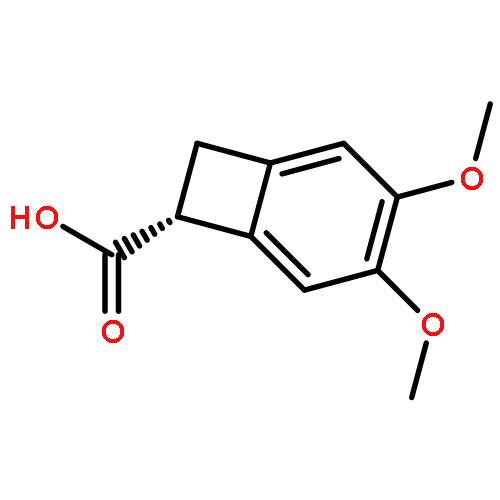
![(S)-3,4-dimethoxy-bicyclo[4.2.0]octa-1,3,5-triene-7-carboxylic acid N-methyl-amide](http://img.cochemist.com/ccimg/1221000/1220993-43-0.png)
![(S)-3,4-dimethoxy-bicyclo[4.2.0]octa-1,3,5-triene-7-carboxylic acid N-methyl-amide](http://img.cochemist.com/ccimg/1221000/1220993-43-0_b.png)
![[1,2,4]TRIAZOLO[1,5-A]PYRAZINE, 5,6,7,8-TETRAHYDRO-2-PHENYL-](http://img.cochemist.com/ccimg/958700/958669-59-5.png)
![[1,2,4]TRIAZOLO[1,5-A]PYRAZINE, 5,6,7,8-TETRAHYDRO-2-PHENYL-](http://img.cochemist.com/ccimg/958700/958669-59-5_b.png)
![PIPERIDINE, 3-[[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]METHYL]-](http://img.cochemist.com/ccimg/876200/876147-50-1.png)
![PIPERIDINE, 3-[[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]METHYL]-](http://img.cochemist.com/ccimg/876200/876147-50-1_b.png)

![(S)-1-(3,4-Dimethoxybicyclo[4.2.0]octa-1,3,5-trien-7-yl)-N-methylmethanamine](http://img.cochemist.com/ccimg/866800/866783-12-2.png)
![(S)-1-(3,4-Dimethoxybicyclo[4.2.0]octa-1,3,5-trien-7-yl)-N-methylmethanamine](http://img.cochemist.com/ccimg/866800/866783-12-2_b.png)
![Thieno[3,2-c]pyridine,2-bromo-4,5,6,7-tetrahydro-, hydrobromide (1:1)](http://img.cochemist.com/ccimg/733800/733757-75-0.png)
![Thieno[3,2-c]pyridine,2-bromo-4,5,6,7-tetrahydro-, hydrobromide (1:1)](http://img.cochemist.com/ccimg/733800/733757-75-0_b.png)
![2-(Trifluoromethyl)-5,6,7,8-tetrahydro[1,2,4]triazolo[1,5-a]pyrazine](http://img.cochemist.com/ccimg/681300/681249-57-0.png)
![2-(Trifluoromethyl)-5,6,7,8-tetrahydro[1,2,4]triazolo[1,5-a]pyrazine](http://img.cochemist.com/ccimg/681300/681249-57-0_b.png)








![tert-Butyl 4,5-dihydrothieno[2,3-c]pyridine-6(7H)-carboxylate](http://img.cochemist.com/ccimg/166000/165947-52-4.png)
![tert-Butyl 4,5-dihydrothieno[2,3-c]pyridine-6(7H)-carboxylate](http://img.cochemist.com/ccimg/166000/165947-52-4_b.png)
![Pyrrolidine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3R)-](http://img.cochemist.com/ccimg/159400/159330-31-1.png)
![Pyrrolidine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3R)-](http://img.cochemist.com/ccimg/159400/159330-31-1_b.png)


![2-(Trifluoromethyl)-5,6,7,8-tetrahydroimidazo[1,2-a]pyrazine](http://img.cochemist.com/ccimg/126100/126069-70-3.png)
![2-(Trifluoromethyl)-5,6,7,8-tetrahydroimidazo[1,2-a]pyrazine](http://img.cochemist.com/ccimg/126100/126069-70-3_b.png)
![4,5,6,7-Tetrahydro-[1,2,3]triazolo[1,5-a]pyrazine](http://img.cochemist.com/ccimg/123300/123291-54-3.png)
![4,5,6,7-Tetrahydro-[1,2,3]triazolo[1,5-a]pyrazine](http://img.cochemist.com/ccimg/123300/123291-54-3_b.png)
![2-(Trifluoromethyl)imidazo[1,2-a]pyrazine](http://img.cochemist.com/ccimg/109200/109113-96-4.png)
![2-(Trifluoromethyl)imidazo[1,2-a]pyrazine](http://img.cochemist.com/ccimg/109200/109113-96-4_b.png)
![Piperidine, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/97300/97231-91-9.png)
![Piperidine, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/97300/97231-91-9_b.png)
![Ethyl 5,6,7,8-tetrahydroimidazo[1,2-a]pyrazine-2-carboxylate](http://img.cochemist.com/ccimg/91500/91476-82-3.png)
![Ethyl 5,6,7,8-tetrahydroimidazo[1,2-a]pyrazine-2-carboxylate](http://img.cochemist.com/ccimg/91500/91476-82-3_b.png)

![PYRIDINE, 5-BROMO-2-[[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]METHYL]-](http://img.cochemist.com/ccimg/88200/88139-92-8.png)
![PYRIDINE, 5-BROMO-2-[[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]METHYL]-](http://img.cochemist.com/ccimg/88200/88139-92-8_b.png)
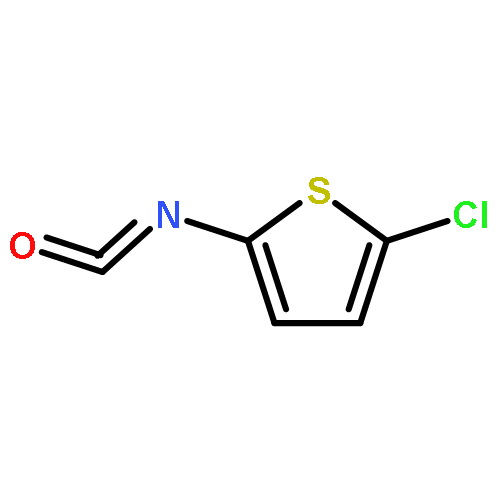
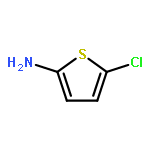
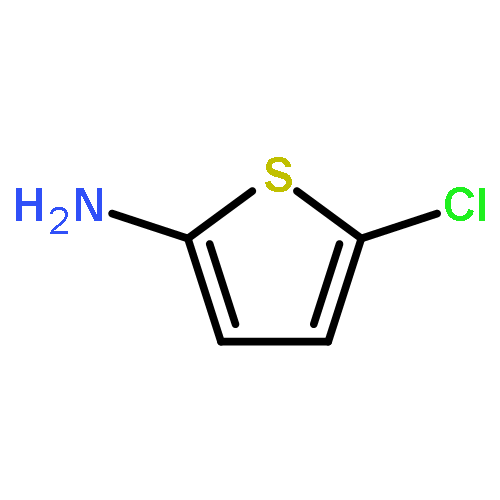


![3,4-Dimethoxybicyclo[4.2.0]octa-1,3,5-triene-7-carboxylic acid](http://img.cochemist.com/ccimg/41300/41234-23-5.png)
![3,4-Dimethoxybicyclo[4.2.0]octa-1,3,5-triene-7-carboxylic acid](http://img.cochemist.com/ccimg/41300/41234-23-5_b.png)
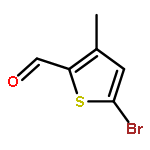
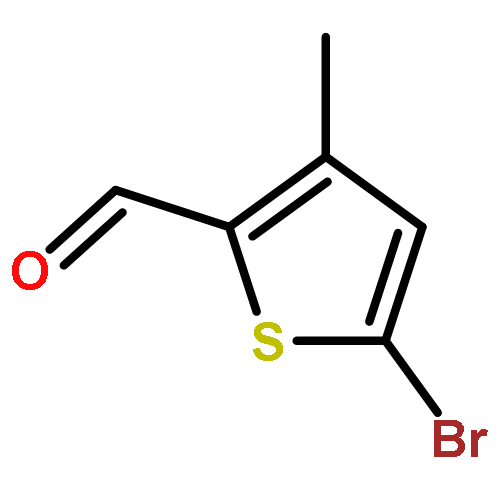
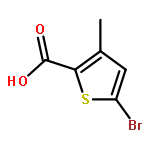
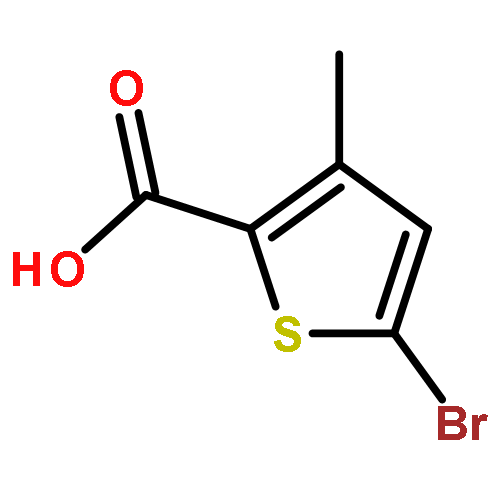


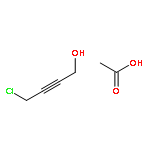
![Acetic acid,2-[(4-chlorophenyl)amino]-2-oxo-](http://img.cochemist.com/ccimg/17800/17738-71-5.png)
![Acetic acid,2-[(4-chlorophenyl)amino]-2-oxo-](http://img.cochemist.com/ccimg/17800/17738-71-5_b.png)


![5-Chlorobenzo[b]thiophene-2-carboxylic acid](http://img.cochemist.com/ccimg/13800/13771-75-0.png)
![5-Chlorobenzo[b]thiophene-2-carboxylic acid](http://img.cochemist.com/ccimg/13800/13771-75-0_b.png)
![2-[benzyl(prop-2-yn-1-yl)amino]ethanol](http://img.cochemist.com/ccimg/13200/13105-78-7.png)
![2-[benzyl(prop-2-yn-1-yl)amino]ethanol](http://img.cochemist.com/ccimg/13200/13105-78-7_b.png)
![Acetic acid,2-[(2-chloroethyl)thio]-](http://img.cochemist.com/ccimg/4400/4332-50-7.png)
![Acetic acid,2-[(2-chloroethyl)thio]-](http://img.cochemist.com/ccimg/4400/4332-50-7_b.png)



![1H-1,2,4-Triazole, 1-[[(2R,3S)-2-(2,4-difluorophenyl)-3-methyloxiranyl]methyl]-](/data/chemimg/1078800/127000-90-2.png)
![1H-1,2,4-Triazole, 1-[[(2R,3S)-2-(2,4-difluorophenyl)-3-methyloxiranyl]methyl]-](/data/chemimg/1078800/127000-90-2_b.png)