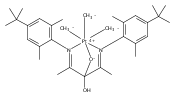
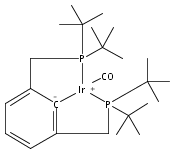
Co-reporter:Jonathan M. Goldberg, Karen I. Goldberg, D. Michael Heinekey, Samantha A. Burgess, David B. Lao, and John C. Linehan
Journal of the American Chemical Society September 13, 2017 Volume 139(Issue 36) pp:12638-12638
Publication Date(Web):September 1, 2017
DOI:10.1021/jacs.7b06480
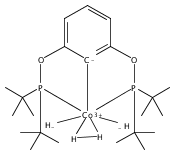
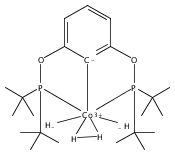
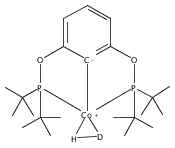
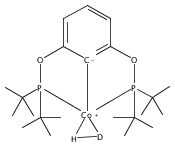
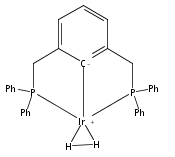
Addition of high pressures of H2 to five-coordinate [(tBu)4(POCOP)Ir(CO)(H)]OTf [(tBu)4(POCOP) = κ3-C6H3-2,6-(OP(tBu)2)2] complexes results in observation of two new iridium–dihydrogen complexes. If the aryl moiety of the POCOP ligand is substituted with an electron withdrawing protonated dimethylamino group at the para position, hydrogen coordination is enhanced. Five-coordinate Ir–H complexes generated by addition of triflic acid to (tBu)4(POCOP)Ir(CO) species show an Ir–H 1H NMR chemical shift dependence on the number of equivalents of acid present. It is proposed that excess triflic acid in solution facilitates triflate dissociation from iridium, resulting in unsaturated five-coordinate Ir–H complexes. The five-coordinate iridium–hydride complexes were found to catalyze H/D exchange between H2 and CD3OD. The existence of the dihydrogen complexes, as well as isotope exchange reactions, provide evidence for proposed ionic hydrogenation intermediates for glycerol deoxygenation.
Co-reporter:Karen I. Goldberg and Alan S. Goldman
Accounts of Chemical Research March 21, 2017 Volume 50(Issue 3) pp:620-620
Publication Date(Web):March 21, 2017
DOI:10.1021/acs.accounts.6b00621
Great progress has been made in the past several decades concerning C–H bond functionalization. But despite many significant advances, a commercially viable large-scale process for selective alkane functionalization remains an unreached goal. Such conversions will require highly active, selective, and long-lived catalysts. In addition, essentially complete atom-economy will be required. Thus, any reagents used in transforming the alkanes must be almost free (e.g., O2, H2O, N2), or they should be incorporated into the desired large-scale product. Any side-products should be completely benign or have value as fuels (e.g., H2 or other alkanes). Progress and promising leads toward the development of such systems involving primarily molecular transition metal catalysts are described.
Co-reporter:Tyler E. Stevens, Karena A. Smoll, and Karen I. Goldberg
Journal of the American Chemical Society June 14, 2017 Volume 139(Issue 23) pp:7725-7725
Publication Date(Web):June 2, 2017
DOI:10.1021/jacs.7b04169
Thermolysis of the RhIII–Me complex (DPEphos)RhMeI2 (1) results in reductive elimination of MeI. Mechanistic studies are consistent with SN2 attack by I− at the RhIII–Me group via two separate competing paths. Addition of sulfur and nitrogen nucleophiles allows effective competition and formation of C(sp3)–S and C(sp3)–N coupled products in high yields. C(sp3)–N bond formation is second-order in amine, consistent with amine substitution of iodide at the metal followed by nucleophilic attack at carbon by a second amine.
Co-reporter:Marie V. Parkes, Wilson D. Bailey, Karen I. Goldberg, Richard A. Kemp
Journal of Organometallic Chemistry 2017 Volume 845(Volume 845) pp:
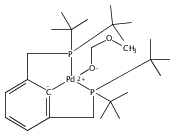
Publication Date(Web):15 September 2017
DOI:10.1016/j.jorganchem.2017.04.026
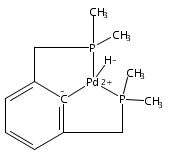
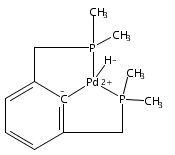
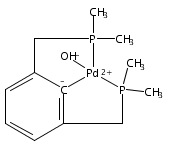
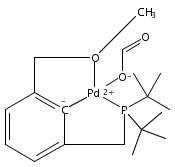
•Hydrogenolysis of five different [XCX]-Pd-OH (X = C,N,O,P,S) complexes examined.•Effects of cis-donor atoms in pincer-Pd-OH complexes studied via DFT.•IES pathway favored over OA/RE pathway.•Hemilabile ligands do not lead to H2 cleavage of the Pd-OH bond.The hydrogenolysis of pincer palladium hydroxides was examined using DFT methods. Two possible mechanistic pathways -- oxidative addition/reductive elimination and internal electrophilic substitution -- were considered for each of five different PdOH complexes that varied in the identity of the donor atoms of the tridentate pincer ligand cis to the hydroxide. For each of the complexes examined, the internal electrophilic substitution pathway was found to be significantly lower in energy than an oxidative addition/reductive elimination pathway. For a PdOH complex bearing a hemilabile pincer ligand, addition of hydrogen resulted in dissociation of the labile arm of the ligand and hydrogenation of the Pd-OH bond to yield a hydride and bound water. This result is consistent with experimental observations for a similar system.A series of pincer-Pd-OH complexes containing strongly-bound cis-donor ligands is shown by DFT to undergo hydrogenolysis via an internal electrophilic substitution pathway. Use of hemilabile cis-ligands does not lead to hydrogenolysis of the Pd-OH bond, but rather hydrogenation to produce a water-bound Pd-H, matching the experimental results.Download high-res image (87KB)Download full-size image
Co-reporter:Jonathan M. Goldberg, Janna L. Berman, Werner Kaminsky, Karen I. Goldberg, D. Michael Heinekey
Journal of Organometallic Chemistry 2017 Volume 845(Volume 845) pp:
Publication Date(Web):15 September 2017
DOI:10.1016/j.jorganchem.2017.04.036
•Formation of stable cis-diiodide iridium carbonyl complexes.•Observation of an oxidative addition intermediate.•I2 addition to square planar Ir(I) complexes likely proceeds via an SN2 mechanism.Reactions of (X)(tBu)4(POCOP)Ir(CO) [(X)(tBu)4(POCOP) = κ3-C6H2-2,6-(OP(tBu)2)2-4-X where X = H, Me, COOMe, OMe, NMe2] complexes with iodine proceed to form Ir(III) iodide carbonyl complexes. When X = H, Me, or COOMe, cis-diiodide carbonyl complexes are observed; however if X = NMe2, a cationic five-coordinate monoiodo carbonyl complex is formed. The stability of the five-coordinate complex is attributed to the unfavorability of iodide coordination to an electron-rich metal center. When X = OMe, iodine addition is proposed to form an equilibrium mixture of a cis-diiodide complex and a five-coordinate monoiodo species. The data are consistent with a mechanism featuring oxidative addition of iodine to Ir(I) to give a cationic monoiodo carbonyl complex, followed by iodide coordination to give a cis-diiodide if the metal center is sufficiently Lewis acidic.Download high-res image (164KB)Download full-size image
Co-reporter:Timothy P. Brewster, Nomaan M. Rezayee, Zuzana Culakova, Melanie S. Sanford, and Karen I. Goldberg
ACS Catalysis 2016 Volume 6(Issue 5) pp:3113
Publication Date(Web):April 15, 2016
DOI:10.1021/acscatal.6b00263
Half-sandwich iridium bipyridine complexes catalyze the hydrogenation of esters and lactones under base-free conditions. The reactions proceed with a variety of ester and lactone substrates. Mechanistic studies implicate a pathway involving rate-limiting hydride transfer to the substrate at high pressures of H2 (≥50 bar).Keywords: acid; ester; homogeneous catalysis; hydrogenation; iridium
Co-reporter:Timothy P. Brewster, Jonathan M. Goldberg, Jeremy C. Tran, D. Michael Heinekey, and Karen I. Goldberg
ACS Catalysis 2016 Volume 6(Issue 9) pp:6302
Publication Date(Web):August 23, 2016
DOI:10.1021/acscatal.6b02130
A family of (para-cymene)RuII complexes are shown to be competent precatalysts for the oxidation of aldehydes to carboxylic acids using water as the oxidant. This reaction, known as the “aldehyde–water shift” (AWS), has been previously demonstrated to be in competition with aldehyde disproportionation. For the few reported mononuclear catalysts for this reaction, either high selectivity for AWS and low conversion or low AWS selectivity and high conversion is observed. A homogeneous precatalyst which is both highly selective for the desired AWS and is highly efficient for conversion of the aldehyde to products is reported herein. In addition, catalyst activity is found to be general to a variety of sterically unencumbered aliphatic aldehydes producing the corresponding carboxylic acid and hydrogen gas.Keywords: aldehyde oxidation; aldehyde−water shift; dehydrogenation; homogeneous catalysis; water
Co-reporter:Jonathan M. Goldberg, Sophia D. T. Cherry, Louise M. Guard, Werner Kaminsky, Karen I. Goldberg, and D. Michael Heinekey
Organometallics 2016 Volume 35(Issue 20) pp:3546-3556
Publication Date(Web):October 7, 2016
DOI:10.1021/acs.organomet.6b00598
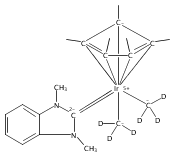
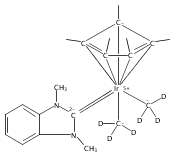
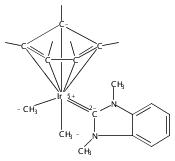
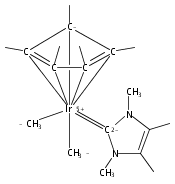
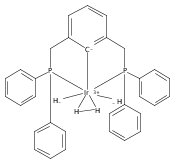
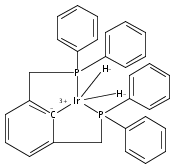
(POCOP)IrI(CO) [POCOP = κ3-C6H3-2,6-(OPR2)2 for R = tBu, iPr] and (PCP)IrI(CO) [PCP = κ3-C6H3-2,6-(CH2PR2)2 for R = tBu and iPr] complexes can add hydrogen via two distinct pathways. When R = tBu, (POCOP)Ir(CO) and (PCP)Ir(CO) complexes only add hydrogen via a proton-catalyzed pathway due to steric effects, yielding trans-dihydride complexes. For R = iPr, both systems oxidatively add hydrogen to give cis-dihydride complexes which thermally isomerize to more thermodynamically favorable trans-dihydride species, consistent with previous reports. Proton-catalyzed hydrogen addition pathways are also observed for both iPr-substituted (pincer)Ir(CO) complexes. (PCP)Ir(CO) complexes add hydrogen under milder conditions than the analogous POCOP species. Intermediate hydrido-pyridine Ir(III) carbonyl complexes from the proton-catalyzed pathway have been synthesized and characterized. This is the first report of a series of complexes shown to add hydrogen via concerted oxidative addition or a proton-catalyzed pathway to the same iridium center.
Co-reporter:Wilson D. Bailey, Lapo Luconi, Andrea Rossin, Dmitry Yakhvarov, Sarah E. Flowers, Werner Kaminsky, Richard A. Kemp, Giuliano Giambastiani, and Karen I. Goldberg
Organometallics 2015 Volume 34(Issue 16) pp:3998-4010
Publication Date(Web):July 31, 2015
DOI:10.1021/acs.organomet.5b00355
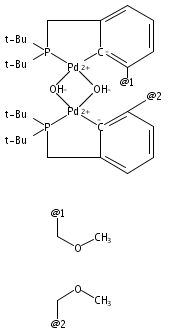
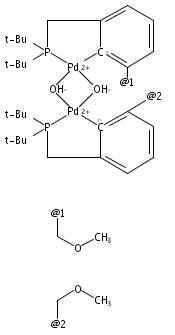
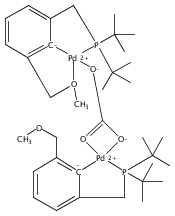
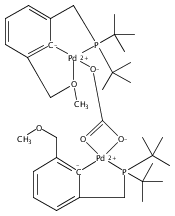
Palladium complexes of the novel unsymmetrical phosphine pyrazole-containing pincer ligands PCNH (PCNH = 1-[3-[(di-tert-butylphosphino)methyl]phenyl]-1H-pyrazole) and PCNMe (PCNMe = 1-[3-[(di-tert-butylphosphino)methyl]phenyl]-5-methyl-1H-pyrazole) have been prepared and characterized through single-crystal X-ray diffraction and multinuclear 1H, 13C{1H}, and 31P{1H} NMR spectroscopy. In preparations of the monomeric hydroxide species (PCNH)Pd(OH), an unexpected N detachment followed by C–H activation on the heterocycle 5-position took place resulting in conversion of the monoanionic {P,C–,N} framework into a dianionic {P,C–,C–} ligand set. The dinuclear hydroxide-bridged species (PCNH)Pd(μ-OH)Pd(PCC) was the final product obtained under ambient conditions. The “rollover” activation was followed via 31P{1H} NMR spectroscopy, and dinuclear cationic μ-OH and monomeric PdII hydroxide intermediates were identified. DFT computational analysis of the process (M06//6-31G*, THF) showed that the energy barriers for the pyrazolyl rollover and for C–H activation through a σ-bond metathesis reaction are low enough to be overcome under ambient-temperature conditions, in line with the experimental findings. In contrast to the PCNH system, no “rollover” reactivity was observed in the PCNMe system, and the terminal hydroxide complex (PCNMe)Pd(OH) could be readily isolated and fully characterized.
Co-reporter:Jonathan M. Goldberg, Gene W. Wong, Kenzie E. Brastow, Werner Kaminsky, Karen I. Goldberg, and D. Michael Heinekey
Organometallics 2015 Volume 34(Issue 4) pp:753-762
Publication Date(Web):February 3, 2015
DOI:10.1021/om501166w
POCOP ligands (POCOP = C6H4-1,3-[OPR2]2) with R= tBu substituents on phosphorus react with Ir(CO)2Cl(p-toluidine) to afford square planar Ir(I) carbonyl complexes. Replacement of tBu groups with iPr groups results in the formation of six-coordinate hydrido-chloride carbonyl complexes of Ir(III). Analogous reactions of POCOP ligands with R groups smaller than iPr require meta-disubstitution in the aromatic ring to prevent precipitation of an insoluble polymer upon metalation with iridium. Structures of five-coordinate cationic hydrido carbonyl species show that a small change in steric demand of the alkyl groups on phosphorus can open the metal coordination sphere, with smaller alkyl groups favoring six-coordinate Ir(III) complexes.
Co-reporter:Margaret L. Scheuermann, Kyle A. Grice, Matthew J. Ruppel, Marta Roselló-Merino, Werner Kaminsky and Karen I. Goldberg
Dalton Transactions 2014 vol. 43(Issue 31) pp:12018-12025
Publication Date(Web):30 Jun 2014
DOI:10.1039/C4DT01143K
The thermolyses of (tBuP(O)N)PtMe2 (1, tBuP(O)N = (di-tert-butylphosphinito)pyridine) and (tBuP(N–H)N)PtMe2 (3, tBuP(N–H)N = (di-tert-butylphosphino)-2-aminopyridine) in benzene-d6 were investigated. With (tBuP(O)N)PtMe2, the product of a rollover cyclometalation of the pyridyl ring was observed in 80% yield along with formation of CH4. In contrast, thermolysis of (tBuP(N–H)N)PtMe2 resulted in competing rollover cyclometalation and intermolecular benzene C–H activation with production of a mixture of CH4 and CH3D.
Co-reporter:Wilson D. Bailey, Werner Kaminsky, Richard A. Kemp, and Karen I. Goldberg
Organometallics 2014 Volume 33(Issue 10) pp:2503-2509
Publication Date(Web):May 5, 2014
DOI:10.1021/om500054f
The synthesis and characterization of anionic, neutral, and cationic hydride complexes of platinum and palladium are reported utilizing the PNP (PNP = 2,6-bis(di-tert-butylphosphinomethyl)pyridine) ligand. Comparisons by IR spectroscopy and X-ray crystallography are made across the series. Evaluation of the metal hydride stretching frequencies of the cationic through anionic complexes shows a trend of increasing M–H bond activation. The reactivity of these metal hydrides with oxygen is evaluated and compared to previously reported oxygen insertion reactions.
Co-reporter:Dr. Margaret L. Scheuermann ; Karen I. Goldberg
Chemistry - A European Journal 2014 Volume 20( Issue 45) pp:14556-14568
Publication Date(Web):
DOI:10.1002/chem.201402599
Abstract
Knowledge of exactly how metal complexes react with molecular oxygen is still limited and this has hampered efforts to develop catalysts for oxidation reactions using O2 as the oxidant and/or oxygen-atom source. A better understanding of the reactions of different types of metal complexes with O2 will be of great utility in rational catalyst development. Reactions between molecular oxygen and Pd0–II and Pt0–IV complexes are reviewed here.
Co-reporter:Marie L. Clement;Kyle A. Grice;Avery T. Luedtke;Werner Kaminsky; Karen I. Goldberg
Chemistry - A European Journal 2014 Volume 20( Issue 52) pp:17287-17291
Publication Date(Web):
DOI:10.1002/chem.201405174
Abstract
PtII complexes containing unsymmetrical (pyridyl)pyrrolide ligands are shown to catalyze the hydroarylation of unactivated alkenes with selectivity for the anti-Markovnikov product. Substitution on the pyrrolide portion of the ligand allows effective tuning of the selectivity to anti-Markovnikov alkylarene products, whereas substitution on the pyridyl portion can promote competitive alkenylarene production.
Co-reporter:Dr. Margaret L. Scheuermann;David W. Boyce; Kyle A. Grice; Werner Kaminsky; Stefan Stoll; William B. Tolman; Ole Swang; Karen I. Goldberg
Angewandte Chemie International Edition 2014 Volume 53( Issue 25) pp:6492-6495
Publication Date(Web):
DOI:10.1002/anie.201402484
Abstract
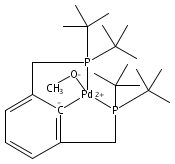
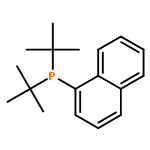
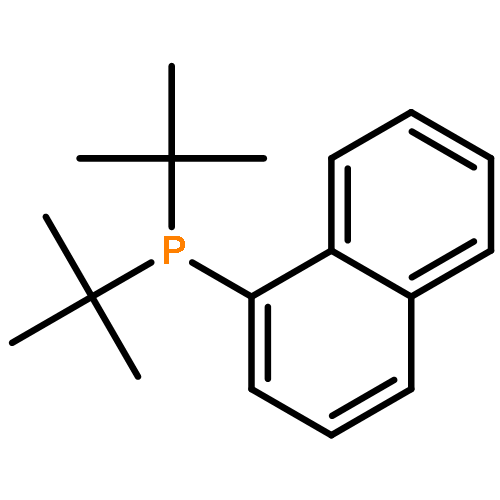
[Pd(P(Ar)(tBu)2)2] (1, Ar=naphthyl) reacts with molecular oxygen to form PdII hydroxide dimers in which the naphthyl ring is cyclometalated and one equivalent of phosphine per palladium atom is released. This reaction involves the cleavage of both CH and OO bonds, two transformations central to catalytic aerobic oxidizations of hydrocarbons. Observations at low temperature suggest the initial formation of a superoxo complex, which then generates a peroxo complex prior to the CH activation step. A transition state for energetically viable CH activation across a Pdperoxo bond was located computationally.
Co-reporter:Kate E. Allen, D. Michael Heinekey, Alan S. Goldman, and Karen I. Goldberg
Organometallics 2014 Volume 33(Issue 6) pp:1337-1340
Publication Date(Web):March 11, 2014
DOI:10.1021/om401241e
(dmPhebox)Ir(OAc)2(OH2) (1a) has previously been found to promote stoichiometric alkane dehydrogenation, affording alkene, acetic acid, and the corresponding Ir hydride complex (dmPhebox)Ir(OAc)(H) (2a) in high yield. In this study, we describe the use of oxygen to quantitatively regenerate 1a from 2a and HOAc. Distinct reaction intermediates are observed during the conversion of 2a to 1a, depending upon the presence or absence of 1 equiv of acetic acid, highlighting potential mechanistic implications regarding alkane dehydrogenation catalysis and the use of oxygen as an oxidant in such systems.
Co-reporter:Jason D. Prantner, Werner Kaminsky, and Karen I. Goldberg
Organometallics 2014 Volume 33(Issue 13) pp:3227-3230
Publication Date(Web):June 19, 2014
DOI:10.1021/om500243n
Reaction of molecular oxygen with the methylplatinum(II) complex K[(κ2-NNO)PtII(CH3)(OH)] (3; NNO = bis(3,5-dimethylpyrazol-1-yl)acetate) in D2O results in oxidation to (κ3-NNO)PtIV(CH3)(OD)2 along with competitive methyl transfer to produce (κ3-NNO)PtIV(CH3)2(OD). Methyl transfer is favored under more alkaline conditions and at higher temperatures. Mechanistic studies are consistent with the direct reaction of 3 with molecular oxygen as a common first reaction step on the path to both products.
Co-reporter:Dr. Margaret L. Scheuermann;David W. Boyce; Kyle A. Grice; Werner Kaminsky; Stefan Stoll; William B. Tolman; Ole Swang; Karen I. Goldberg
Angewandte Chemie 2014 Volume 126( Issue 25) pp:6610-6613
Publication Date(Web):
DOI:10.1002/ange.201402484
Abstract
[Pd(P(Ar)(tBu)2)2] (1, Ar=naphthyl) reacts with molecular oxygen to form PdII hydroxide dimers in which the naphthyl ring is cyclometalated and one equivalent of phosphine per palladium atom is released. This reaction involves the cleavage of both CH and OO bonds, two transformations central to catalytic aerobic oxidizations of hydrocarbons. Observations at low temperature suggest the initial formation of a superoxo complex, which then generates a peroxo complex prior to the CH activation step. A transition state for energetically viable CH activation across a Pdperoxo bond was located computationally.
Co-reporter:Alexander J. M. Miller, Werner Kaminsky, and Karen I. Goldberg
Organometallics 2014 Volume 33(Issue 5) pp:1245-1252
Publication Date(Web):February 20, 2014
DOI:10.1021/om5000166
An arene activation strategy for the selective disassembly of aryl ethers is reported. A variety of aryl ethers readily bind an electrophilic pentamethylcyclopentadienyl iridium center by η6-arene coordination, generating complexes that are activated toward hydrolysis and cleavage of the Ar–OR bond (R = Me, Et, Ph). Hydrolysis occurs rapidly at room temperature in aqueous pH 7 phosphate buffer (or upon modest heating under acidic conditions), releasing alcohol while forming cyclohexadienyl-one products. Under strongly acidic conditions, protonation of the dienyl-one followed by substitution with starting aryl ether completes a hydrolysis cycle. Mechanistic studies suggest that the key hydrolysis step proceeds via nucleophilic attack at the ipso position of the arene (SNAr mechanism). The observed mechanism is considered in the context of lignocellulosic biomass conversion.
Co-reporter:Timothy P. Brewster ; Alexander J. M. Miller ; D. Michael Heinekey
Journal of the American Chemical Society 2013 Volume 135(Issue 43) pp:16022-16025
Publication Date(Web):October 21, 2013
DOI:10.1021/ja408149n
A series of half-sandwich Ir and Rh compounds are demonstrated to be competent catalysts for the hydrogenation of carboxylic acids under relatively mild conditions. Of the structurally diverse group of catalysts tested for activity, a Cp*Ir complex supported by an electron-releasing 2,2′-bipyridine ligand was the most active. Higher activity was achieved with employment of Brønsted or Lewis acid promoters. Mechanistic studies suggest a possible reaction pathway involving activated carboxylic acid substrates. The hydrogenation reaction was shown to be general to a variety of aliphatic acids.
Co-reporter:David B. Lao, Alisa C. E. Owens, D. Michael Heinekey, and Karen I. Goldberg
ACS Catalysis 2013 Volume 3(Issue 10) pp:2391
Publication Date(Web):September 5, 2013
DOI:10.1021/cs400551g
Iridium pincer complexes (POCOP)Ir(CO) (POCOP = κ3-C6H3-1,3-[OP(tBu)2]2) and substituted POCOP derivatives catalyze deoxygenation of glycerol to n-propanol and 1,3-propanediol in good yield under moderate conditions (acidic aqueous dioxane, 200 °C, 80 bar H2). Catalyst solubility in the polar reaction mixture is improved by incorporation of a polar moiety in the para position of the POCOP phenyl ring, with the best results obtained with a dimethylamino substituent.Keywords: catalysis; deoxygenation; glycerol
Co-reporter: Alexer J. M. Miller; D. Michael Heinekey; James M. Mayer; Karen I. Goldberg
Angewandte Chemie International Edition 2013 Volume 52( Issue 14) pp:3981-3984
Publication Date(Web):
DOI:10.1002/anie.201208470
Co-reporter:Kate E. Allen, D. Michael Heinekey, Alan S. Goldman, and Karen I. Goldberg
Organometallics 2013 Volume 32(Issue 6) pp:1579-1582
Publication Date(Web):February 18, 2013
DOI:10.1021/om301267c
Stoichiometric alkane dehydrogenation utilizing an IrIII pincer complex, (dmPhebox)Ir(OAc)2(OH2) (1a), has been described. The reaction between 1a and octane resulted in quantitative formation of (dmPhebox)Ir(OAc)(H) (3a) and octene. At early reaction times 1-octene is the major product, indicative of terminal C–H activation by 1a. In contrast to prior reports of alkane dehydrogenation with Ir, C–H bond activation occurs at IrIII and the dehydrogenation is not inhibited by nitrogen, olefin, or water.
Co-reporter:Margaret L. Scheuermann, Avery T. Luedtke, Susan K. Hanson, Ulrich Fekl, Werner Kaminsky, and Karen I. Goldberg
Organometallics 2013 Volume 32(Issue 17) pp:4752-4758
Publication Date(Web):August 26, 2013
DOI:10.1021/om4003363
The reactions of various five-coordinate Pt(IV) complexes with molecular oxygen have been studied. In arene solution, the complexes (tBuMe2nacnac)PtMe3 (1; tBuMe2nacnac– = [((4-tBu-2,6-Me2C6H2)NC(CH3))2CH]−), (Me3Me-nacnac)PtMe3 (2; Me3Me-nacnac– = [((2,4,6-Me3C6H2)NC(CH3))2CCH3]−), and (tBu2PyPyr)PtMe3 (3; tBu2PyPyr– = 3,5-di-tert-butyl-2-(2-pyridyl)pyrrolide) reacted immediately with oxygen to form peroxo species in which two oxygen atoms bridge between the metal center and a carbon atom in the ligand backbone. In contrast, no reaction between (iPr2AnIm)PtMe3 (4a; iPr2AnIm– = [o-C6H4-{N(C6H3iPr2)}(CH═NC6H3iPr2)]−) or (Me3AnIm)PtMe3 (4b; Me3AnIm– = [o-C6H4-{N(C6H2Me3)}(CH═NC6H2Me3)]−) and oxygen was observed. As activation of oxygen by five-coordinate Pt(IV) species was found to involve cooperation between the metal center and the ligand, the ability of the ligand to participate in the oxygen binding appears to be a vital component. Oxygen atom transfer reactions of the novel peroxo species are also presented.
Co-reporter:Luc Boisvert and Karen I. Goldberg
Accounts of Chemical Research 2012 Volume 45(Issue 6) pp:899
Publication Date(Web):May 11, 2012
DOI:10.1021/ar2003072
Limited natural resources, high energy consumption, economic considerations, and environmental concerns demand that we develop new technologies for the sustainable production of chemicals and fuels. New methods that combine the selective activation of C–H bonds of hydrocarbons with oxidation by a green oxidant such as molecular oxygen would represent huge advances toward this goal. The spectacular selectivity of transition metals in cleaving C–H bonds offers the potential for the direct use of hydrocarbons in the production of value-added organics such as alcohols. However, the use of oxygen, which is abundant, environmentally benign, and inexpensive (particularly from air), has proven challenging, and more expensive and less green oxidants are often employed in transition-metal-catalyzed reactions. Advances in the use of oxygen as an oxidant in transition-metal-catalyzed transformations of hydrocarbons will require a better understanding of how oxygen reacts with transition metal alkyl and hydride complexes. For alkane oxidations, researchers will need to comprehend and predict how metals that have shown particularly high activity and selectivity in C–H bond activation (e.g. Pt, Pd, Rh, Ir) will react with oxygen.In this Account, we present our studies of reactions of late metal alkyls and hydrides with molecular oxygen, emphasizing the mechanistic insights that have emerged from this work. Our studies have unraveled some of the general mechanistic features of how molecular oxygen inserts into late metal hydride and alkyl bonds along with a nascent understanding of the scope and limitations of these reactions. We present examples of the formation of metal hydroperoxide species M-OOH by insertion of dioxygen into Pt(IV)–H and Pd(II)–H bonds and show evidence that these reactions proceed by radical chain and hydrogen abstraction pathways, respectively. Comparisons with recent reports of insertion of oxygen into other Pd(II)–H complexes, and also into Ir(III)–H and Rh(III)–H complexes, point to potentially general mechanisms for this type of reaction.Additionally, we observed oxygen-promoted C–H and H–H reductive elimination reactions from five-coordinate Ir(III) alkyl hydride and dihydride complexes, respectively. Further, when Pd(II)Me2 and Pt(II)Me2 complexes were exposed to oxygen, insertion processes generated M–OOMe complexes. Mechanistic studies for these reactions are consistent with radical chain homolytic substitution pathways involving five-coordinate M(III) intermediates. Due to the remarkable ability of Pt(II) and Pd(II) to activate the C–H bonds of hydrocarbons (RH) and form M–R species, this reactivity is especially exciting for the development of partial alkane-oxidation processes that utilize molecular oxygen.Our understanding of how late transition metal alkyls and hydrides react with molecular oxygen is growing rapidly and will soon approach our knowledge of how other small molecules such as olefins and carbon monoxide react with these species. Just as advances in understanding olefin and CO insertion reactions have shaped important industrial processes, key insight into oxygen insertion should lead to significant gains in sustainable commercial selective oxidation catalysis.
Co-reporter:Takiya J. Ahmed Foskey, D. Michael Heinekey, and Karen I. Goldberg
ACS Catalysis 2012 Volume 2(Issue 6) pp:1285
Publication Date(Web):May 1, 2012
DOI:10.1021/cs300120d
Iridium pincer complex (POCOP)IrH2 (1; POCOP = κ3-C6H3-1,3-[OP(tBu)2]2) catalyzes hydrogenolysis of 1,2-propanediol to n-propanol in good yield under mild conditions (acidic aqueous dioxane, 50–125 °C, 100–600 psi H2). Studies of catalyst speciation revealed that the catalyst reservoir species is (POCOP)Ir(CO) (2), formed by decarbonylation of the substrate. Complex 2 is a superior catalyst precursor, since it is air-stable and readily prepared by treating complex 1 with CO.Keywords: catalysis; diol deoxygenation; hydrogenation; hydrogenolysis;
Co-reporter:Gregory R. Fulmer ; Alexandra N. Herndon ; Werner Kaminsky ; Richard A. Kemp
Journal of the American Chemical Society 2011 Volume 133(Issue 44) pp:17713-17726
Publication Date(Web):September 21, 2011
DOI:10.1021/ja205824q
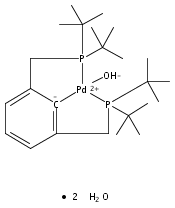
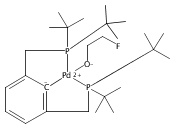
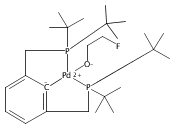
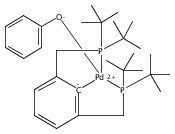
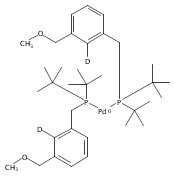
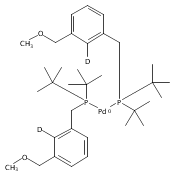
A series of pincer (tBuPCP)Pd(II)–OR complexes (tBuPCP = 2,6-bis(CH2PtBu2)C6H3, R = H, CH3, C6H5, CH2C(CH3)3, CH2CH2F, CH2CHF2, CH2CF3) were synthesized to explore the generality of hydrogenolysis reactions of palladium–oxygen bonds. Hydrogenolysis of the Pd hydroxide complex to generate the Pd hydride complex and water was shown to be inhibited by formation of a water-bridged, hydrogen-bonded Pd(II) hydroxide dimer. The Pd alkoxide and aryloxide complexes exhibited more diverse reactivity. Depending on the characteristics of the −OR ligand (steric bulk, electron-donating ability, and/or the presence of β-hydrogen atoms), hydrogenolysis was complicated by hydrolysis by adventitious water, a lack of reactivity with hydrogen, or a competing dissociative β-hydride abstraction reaction pathway. Full selectivity for hydrogenolysis was observed with the partially fluorinated Pd(II) 2-fluoroethoxide complex. The wide range of Pd–OR substrates examined helps to clarify the variety of reaction pathways available to late-transition-metal alkoxides as well as the conditions necessary to tune the reactivity to hydrogenolysis, hydrolysis, or dissociative β-hydride abstraction.
Co-reporter:Raymond B. Lansing, Karen I. Goldberg and Richard A. Kemp
Dalton Transactions 2011 vol. 40(Issue 35) pp:8950-8958
Publication Date(Web):20 May 2011
DOI:10.1039/C1DT10265F
Two new unsymmetrical RPNPR′-type pincer ligands based on a bis(tolyl)amine framework have been synthesized and characterized by a variety of techniques, including X-ray crystallography. These ligands have been coordinated to Ni, Pd, and Pt precursors to provide a number of well-characterized group 10 halides. Conversion of these metal halides to metal hydrides was accomplished using borohydride reagents, or by direct interaction of the ligand with the zerovalent metal precursor. The insertion of oxygen into these hydrides in an attempt to prepare metal hydroperoxides has been examined; however, we were unable to obtain stable and isolable hydroperoxide species.
Co-reporter:Kyle A. Grice, Werner Kaminsky, Karen I. Goldberg
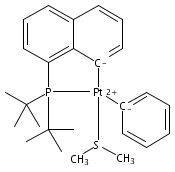
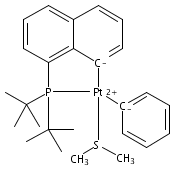
Inorganica Chimica Acta 2011 Volume 369(Issue 1) pp:76-81
Publication Date(Web):15 April 2011
DOI:10.1016/j.ica.2010.11.021
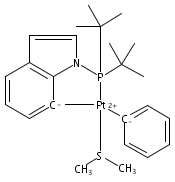
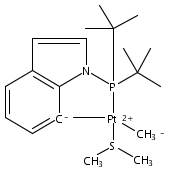
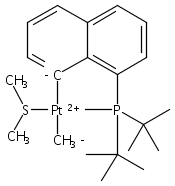
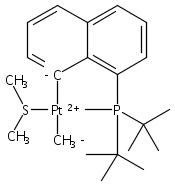
The bulky phosphine ligands di-tert-butyl(1-naphthyl)phosphine (1) or di-tert-butyl(N-indolyl)phosphine (2) react at room temperature with [(μ-SMe2)PtMe2]2. Coordination of the phosphine and C–H bond activation at an sp2 carbon of the ligand with the release of methane takes place to form the PC cyclometalated products [(PC)PtMe(SMe2)] (3 or 4, respectively). The cyclometalated complexes 3 and 4 have both been characterized by X-ray crystallography. Complexes 3 and 4 were each observed to undergo intermolecular activation of arene C–H bonds. Upon thermolysis in benzene, complexes 3 and 4 react to eliminate methane and yield isolable platinum(II)–phenyl complexes.Graphical abstractTwo bulky phosphine ligands react at room temperature with [(μ-SMe2)PtMe2]2 to form the PC cyclometalated products (PC)PtMe(SMe2). The cyclometalated complexes have both been characterized by X-ray crystallography. Pt–Ph products resulting from intermolecular C–H activation were observed upon thermolysis of the cyclometalated Pt–Me complexes in benzene.Research highlights► Pt(II)–Me and Ph complexes bearing cyclometalated phosphine ligands. ► Structural characterization of Pt(II)–Me complexes of cyclometalated phosphine ligands. ► Intermolecular arene C–H bond activation by Pt(II) phosphine complexes.
Co-reporter:Gregory R. Fulmer, Werner Kaminsky, Richard A. Kemp, and Karen I. Goldberg
Organometallics 2011 Volume 30(Issue 6) pp:1627-1636
Publication Date(Web):March 3, 2011
DOI:10.1021/om101150y
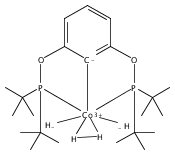
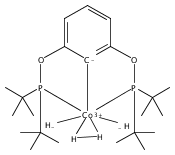
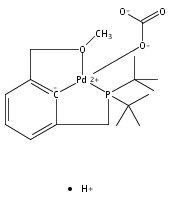
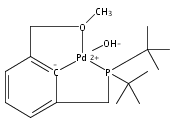
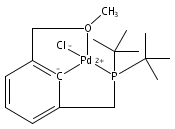
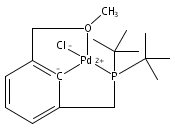
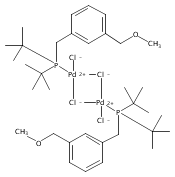
The synthesis of a new pincer ligand (tBuPCO = 2-(CH2PtBu2)-6-(CH2OCH3)C6H3) is reported. This ligand has been observed to coordinate in three different modes to palladium. The tBuPCO ligand coordinates in a monodentate fashion through the phosphine moiety in the dimeric [(tBuPCO)Pd(Cl)(μ-Cl)]2. Bidentate coordination is observed through the phosphine and the aryl ring in the binuclear [(tBuPCO)Pd(μ-OH)]2. The traditional tridentate coordination mode of a pincer is observed in the monomeric complex (tBuPCO)PdCl, wherein the ether oxygen provides the third point of attachment. Each of these novel palladium(II) complexes was characterized by NMR spectroscopy, elemental analyses, and single-crystal X-ray crystallography. A variety of other palladium(II) complexes of tBuPCO have also been prepared and characterized, including the hydroxide complex (tBuPCO)PdOH. The reactivity of the hydroxide complex with CO2, CO, and H2 is reported.
Co-reporter:Abby R. O’Connor, Werner Kaminsky, D. Michael Heinekey, and Karen I. Goldberg
Organometallics 2011 Volume 30(Issue 8) pp:2105-2116
Publication Date(Web):March 29, 2011
DOI:10.1021/om1009473
The synthesis and characterization of (COD)Rh(I) and (NBD)Rh(I) (COD = cyclooctadiene; NBD = norbornadiene) chloride complexes containing the 2-(dicyclohexylphosphino)biphenyl (PCy2biPh) ligand are reported. Abstraction of the halide with Na(BArF)4 yields cationic Rh(I) complexes [(NBD)Rh(PCy2biPh)][B(ArF)4] (2) and [(COD)Rh(PCy2biPh)][B(ArF)4] (7) (ArF = 3,5-bis(trifluoromethyl)phenyl). In complex 2, the pendent arene of the ligand is coordinated in an η2-fashion to rhodium. Complex 7 exists in two configurations that were characterized by low-temperature NMR spectroscopy. One structure is analogous to 2 with η2-coordination of the arene, and the other exhibits η6-coordination. These structures interconvert on the NMR time scale at room temperature. Addition of H2 to complex 2 yields the Rh(III) dihydride complex [(PCy2biPh)RhH2][B(ArF)4] (5), while the addition of H2 to 7 generates the Rh(I) olefin complex [(COE)Rh(PCy2biPh)][B(ArF)4] (8). In both 5 and 8, the pendent arene of the ligand is bound η6 to Rh. Benzene hydrogenation to cyclohexane using 2 as a catalyst precursor is described. Poisoning experiments indicate that heterogeneous rhodium is likely to be the active catalyst in this arene hydrogenation reaction.
Co-reporter:Margaret L. Scheuermann, Ulrich Fekl, Werner Kaminsky, and Karen I. Goldberg
Organometallics 2010 Volume 29(Issue 21) pp:4749-4751
Publication Date(Web):September 27, 2010
DOI:10.1021/om1003946
The Pt(IV) complex (tBuMe2nacnac)PtMe3 (1, tBuMe2nacnac = [((4-tBu-2,6-Me2C6H2)NC(CH3))2CH]) reacts with molecular oxygen to form a peroxo complex, 2, in which one oxygen atom is bound to the platinum center and the other to the central carbon of the nacnac ligand backbone. Over time, 2 converts to a new Pt(IV) species (3) wherein the oxygen−oxygen bond has been cleaved.
Co-reporter:Wesley H. Bernskoetter ; Susan Kloek Hanson ; Sara K. Buzak ; Zoe Davis ; Peter S. White ; Rodney Swartz ; Karen I. Goldberg ;Maurice Brookhart
Journal of the American Chemical Society 2009 Volume 131(Issue 24) pp:8603-8613
Publication Date(Web):June 2, 2009
DOI:10.1021/ja901706b
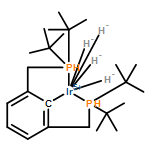
New iridium complexes of a tridentate pincer ligand, 2,6-bis(di-tert-butylphosphinito)pyridine (PONOP), have been prepared and used in the study of hydrocarbon C−H bond activation. Intermolecular oxidative addition of a benzene C−H bond was directly observed with [(PONOP)IrI(cyclooctene)][PF6] at ambient temperature, resulting in a cationic five-coordinate iridium(III) phenyl hydride product. Protonation of the (PONOP)IrI methyl complex yielded the corresponding iridium(III) methyl hydride cation, a rare five-coordinate, 16-valence electron transition metal alkyl hydride species which was characterized by X-ray diffraction. Kinetic studies of C−H bond coupling and reductive elimination reactions from the five-coordinate complexes have been carried out. Exchange NMR spectroscopy measurements established a barrier of 17.8(4) kcal/mol (22 °C) for H−Caryl bond coupling in the iridium(III) phenyl hydride cation and of 9.3(4) kcal/mol (−105 °C) for the analogous H−Calkyl coupling in the iridium(III) methyl hydride cation. The origin of the higher barrier of H−Caryl relative to H−Calkyl bond coupling is proposed to be influenced by a hindered rotation about the Ir−Caryl bond, a result of the sterically demanding PONOP ligand.
Co-reporter:Luc Boisvert ; Melanie C. Denney ; Susan Kloek Hanson
Journal of the American Chemical Society 2009 Volume 131(Issue 43) pp:15802-15814
Publication Date(Web):October 14, 2009
DOI:10.1021/ja9061932
The reaction of (bipy)PdMe2 (1) (bipy = 2,2′-bipyridine) with molecular oxygen results in the formation of the palladium(II) methylperoxide complex (bipy)PdMe(OOMe) (2). The identity of the product 2 has been confirmed by independent synthesis. Results of kinetic studies of this unprecedented oxygen insertion reaction into a palladium alkyl bond support the involvement of a radical chain mechanism. Reproducible rates, attained in the presence of the radical initiator 2,2′-azobis(2-methylpropionitrile) (AIBN), reveal that the reaction is overall first-order (one-half-order in both [1] and [AIBN], and zero-order in [O2]). The unusual rate law (half-order in [1]) implies that the reaction proceeds by a mechanism that differs significantly from those for organic autoxidations and for the recently reported examples of insertion of O2 into Pd(II) hydride bonds. The mechanism for the autoxidation of 1 is more closely related to that found for the autoxidation of main group and early transition metal alkyl complexes. Notably, the chain propagation is proposed to proceed via a stepwise associative homolytic substitution at the Pd center of 1 with formation of a pentacoordinate Pd(III) intermediate.
Co-reporter:Jennifer L. Look ; Douglas D. Wick ; James M. Mayer
Inorganic Chemistry 2009 Volume 48(Issue 4) pp:1356-1369
Publication Date(Web):January 21, 2009
DOI:10.1021/ic801216r
The platinum(IV) hydride complexes TpMe2PtR2H (TpMe2 = hydridotris(3,5-dimethylpyrazolyl)borate, R = Me (1a), Ph (1b)) react with molecular oxygen to form platinum(IV) hydroperoxide complexes TpMe2PtR2OOH (R = Me (2a) and Ph (2b), respectively) in high yield. The results of kinetic and mechanistic studies of these reactions are consistent with the net insertion of molecular oxygen into the Pt(IV)−H bonds occurring via radical chain mechanisms. The radical chain pathways resemble, in many respects, those documented for autoxidations of organic substrates, but significant differences are also evident. The autoxidations of 1a and 1b both autoaccelerate, but the nature of the rate accelerations and the dependence of the rates on the hydroperoxide products are not the same. The different rate laws observed for the reactions of TpMe2PtR2H complexes with molecular oxygen can be rationalized on the basis of similar initiation and propagation events with different chain termination steps.
Co-reporter:Nicole A. Smythe, Kyle A. Grice, B. Scott Williams and Karen I. Goldberg
Organometallics 2009 Volume 28(Issue 1) pp:277-288
Publication Date(Web):November 26, 2008
DOI:10.1021/om800905q
The platinum(IV) hydroxide and methoxide complexes fac-(dppbz)PtMe3(OR) (dppbz = o-bis(diphenylphosphino)benzene; R = H (1), CH3 (2)) have been prepared and characterized. Thermolysis of hydroxide 1 produces (dppbz)PtMe2 (3) and methanol in a rare example of directly observed sp3 carbon−oxygen reductive elimination from a metal center to form an alcohol. Competitive carbon−carbon reductive elimination to form (dppbz)PtMe(OH) (5) and ethane also occurs. In contrast, the major reaction observed upon thermolysis of the methoxide analog 2 is neither carbon−oxygen nor carbon−carbon reductive elimination. Instead, products expected from formal β-hydride elimination followed by carbon−hydrogen reductive elimination are detected. Mechanistic studies suggest the operation of an alternative mechanism to that most commonly accepted for this fundamental reaction; a dissociative β-hydride abstraction pathway is proposed.
Co-reporter:Kyle A. Grice and Karen I. Goldberg
Organometallics 2009 Volume 28(Issue 4) pp:953-955
Publication Date(Web):January 20, 2009
DOI:10.1021/om8011272
The platinum(II) complex (PN)PtMe2 (1; PN = 2-((di-tert-butylphosphino)methyl)pyridine) reacts with molecular oxygen in benzene or methylene chloride solutions to form (PN)PtMe(OOMe) (2), a platinum(II) methylperoxo complex. The structure of the methylperoxo species 2 was determined by X-ray crystallography.
Co-reporter:Brandon L. Dietrich ; Karen I. Goldberg ; D. Michael Heinekey ; Tom Autrey ;John C. Linehan
Inorganic Chemistry 2008 Volume 47(Issue 19) pp:8583-8585
Publication Date(Web):September 12, 2008
DOI:10.1021/ic801161g
Dehydrogenation of amine boranes is catalyzed efficiently by the iridium pincer complex (κ3-1,3-(OPtBu2)2C6H3)Ir(H)2 (1). With CH3NH2BH3 (MeAB) and with AB/MeAB mixtures (AB = NH3BH3), the rapid release of 1 equiv of H2 is observed to yield soluble oligomeric products at rates similar to those previously reported for the dehydrogenation of AB catalyzed by 1. ΔH for the dehydrogenation of AB, MeAB, and AB/MeAB mixtures has been determined by calorimetry. The experimental heats of reaction are compared to results from computational studies.
Co-reporter:Susan Kloek Hanson, D. Michael Heinekey and Karen I. Goldberg
Organometallics 2008 Volume 27(Issue 7) pp:1454-1463
Publication Date(Web):March 4, 2008
DOI:10.1021/om7012259
New rhodium(I) complexes (PNP)Rh(X) (PNP = 2,6-bis(di-tert-butylphosphinomethyl)pyridine) (X = OTf (1), OAc (3), OH (8), OCH2CF3 (9), OC6H5 (10), OC6H4NO2 (11)) have been prepared. Hydroxide complex 8 and trifluoroethoxide complex 9 undergo stoichiometric activation of benzene-d6 to form the phenyl complex (PNP)Rh(C6D5). Acetate and aryloxide complexes 3, 10, and 11 are active catalysts for H−D exchange between arenes and water. Control experiments indicate that the rhodium complexes are the active catalysts and that the observed exchange is not catalyzed by adventitious acid. Mechanistic studies of the H−D exchange reaction support a pathway involving dissociation of aryloxide or acetate ligand. The reaction is accelerated by added alcohol and, for the acetate complex, inhibited by added sodium acetate.
Co-reporter:AveryT. Luedtke ;KarenI. Goldberg
Angewandte Chemie International Edition 2008 Volume 47( Issue 40) pp:7694-7696
Publication Date(Web):
DOI:10.1002/anie.200800524
Co-reporter:AveryT. Luedtke ;KarenI. Goldberg
Angewandte Chemie 2008 Volume 120( Issue 40) pp:7808-7810
Publication Date(Web):
DOI:10.1002/ange.200800524
Co-reporter:Susan M. Kloek;D. Michael Heinekey ;Karen I. Goldberg
Angewandte Chemie 2007 Volume 119(Issue 25) pp:
Publication Date(Web):10 MAY 2007
DOI:10.1002/ange.200700270
Rhodium(I)-Komplexe der Form [(PNP)Rh(OR)] (R=H, CH2CF3, C6H5; PNP=2,6-Bis[(di-tert-butylphosphanyl)methyl]pyridin) wurden hergestellt. Die Hydroxid- und Trifluorethoxidkomplexe aktivieren Benzol bei der Thermolyse in [D6]Benzol. [(PNP)Rh(OC6H5)] katalysiert den H/D-Austausch zwischen D2O und Benzol sowie zwischen H2O und [D8]Toluol (siehe Schema), bei dem der Austausch selektiv an den meta- und para-Positionen verläuft.
Co-reporter:Susan M. Kloek;D. Michael Heinekey ;Karen I. Goldberg
Angewandte Chemie International Edition 2007 Volume 46(Issue 25) pp:
Publication Date(Web):10 MAY 2007
DOI:10.1002/anie.200700270
Rhodium(I) complexes of the form [(PNP)Rh(OR)] (R=H, CH2CF3, C6H5; PNP=2,6-bis[(di-tert-butylphosphino)methyl]pyridine) have been prepared. Upon thermolysis in [D6]benzene, the hydroxide and trifluoroethoxide complexes undergo benzene activation. [(PNP)Rh(OC6H5)] is an active catalyst for the H/D exchange between D2O and benzene and between H2O and [D8]toluene (see scheme), for which exchange occurs selectively at the meta and para positions.
Co-reporter:Margaret L. Scheuermann, Kyle A. Grice, Matthew J. Ruppel, Marta Roselló-Merino, Werner Kaminsky and Karen I. Goldberg
Dalton Transactions 2014 - vol. 43(Issue 31) pp:NaN12025-12025
Publication Date(Web):2014/06/30
DOI:10.1039/C4DT01143K
The thermolyses of (tBuP(O)N)PtMe2 (1, tBuP(O)N = (di-tert-butylphosphinito)pyridine) and (tBuP(N–H)N)PtMe2 (3, tBuP(N–H)N = (di-tert-butylphosphino)-2-aminopyridine) in benzene-d6 were investigated. With (tBuP(O)N)PtMe2, the product of a rollover cyclometalation of the pyridyl ring was observed in 80% yield along with formation of CH4. In contrast, thermolysis of (tBuP(N–H)N)PtMe2 resulted in competing rollover cyclometalation and intermolecular benzene C–H activation with production of a mixture of CH4 and CH3D.
Co-reporter:Raymond B. Lansing, Karen I. Goldberg and Richard A. Kemp
Dalton Transactions 2011 - vol. 40(Issue 35) pp:NaN8958-8958
Publication Date(Web):2011/05/20
DOI:10.1039/C1DT10265F
Two new unsymmetrical RPNPR′-type pincer ligands based on a bis(tolyl)amine framework have been synthesized and characterized by a variety of techniques, including X-ray crystallography. These ligands have been coordinated to Ni, Pd, and Pt precursors to provide a number of well-characterized group 10 halides. Conversion of these metal halides to metal hydrides was accomplished using borohydride reagents, or by direct interaction of the ligand with the zerovalent metal precursor. The insertion of oxygen into these hydrides in an attempt to prepare metal hydroperoxides has been examined; however, we were unable to obtain stable and isolable hydroperoxide species.
.jpg)



![Phosphine,1,1'-[(1,3-phenylene)bis(methylene)]bis[1,1-bis(1-methylethyl)-](http://img.cochemist.com/ccimg/193100/193084-64-9.png)
![Phosphine,1,1'-[(1,3-phenylene)bis(methylene)]bis[1,1-bis(1-methylethyl)-](http://img.cochemist.com/ccimg/193100/193084-64-9_b.png)




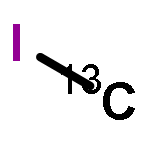



















































































































































































































![[2,2'-Bipyridine]-4,4'-diol](/data/chemimg/1008300/90770-88-0.png)
![[2,2'-Bipyridine]-4,4'-diol](/data/chemimg/1008300/90770-88-0_b.png)


![[Ir(η2-ethylene)2Cl]2](http://img.cochemist.com/ccimg/39800/39722-81-1.png)
![[Ir(η2-ethylene)2Cl]2](http://img.cochemist.com/ccimg/39800/39722-81-1_b.png)





![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)