Co-reporter:Quaovi H. Sodji, James R. Kornacki, John F. McDonald, Milan Mrksich, Adegboyega K. Oyelere
European Journal of Medicinal Chemistry 2015 Volume 96() pp:340-359
Publication Date(Web):26 May 2015
DOI:10.1016/j.ejmech.2015.04.014
•Folate γ-hydroxamate is devoid of HDAC inhibition.•Pteroic hydroxamate is selective against HDAC6, though active against other isoforms such as HDAC1 and 8.•HDACi with pteroic acid (cap group) and hydroxamate (ZBG) are potent against HDAC1 and 6.•Pteroic based-hydroxamates had anticancer activity against FR(+) tumor cells (KB and HeLa).•HDAC1 inhibition results in KB cells death, whereas, HDAC6 does not.Histone deacetylase (HDAC) inhibition has recently emerged as a novel therapeutic approach for the treatment of various pathological conditions including cancer. Currently, two HDAC inhibitors (HDACi) – Vorinostat and Romidepsin – have been approved for the treatment of cutaneous T-cell lymphoma. However, HDACi remain ineffective against solid tumors and are associated with adverse events including cardiotoxicity. Targeted delivery may enhance the therapeutic indices of HDACi and enable them to be efficacious against solid tumors. We showed herein that morphing of folic and pteroic acids into the surface recognition group of HDACi results in hydroxamate and benzamide HDACi which derived tumor homing by targeting folate receptor (FR), a receptor commonly overexpressed in solid tumors. We observed a correlation between the potency of HDAC1 inhibition and cytotoxicity as only the potent pteroate hydroxamates, 11d and 11e, displayed antiproliferative activity against two representative FR-expression cancer cells. Our observation further supports the previous results which suggest that for a drug to be successfully targeted using the FR, it must be extremely potent against its primary target as the FR has a low delivery efficiency.
Co-reporter:Subhasish Tapadar, Shaghayegh Fathi, Idris Raji, Wilson Omesiete, James R. Kornacki, Sandra C. Mwakwari, Masanori Miyata, Kazunori Mitsutake, Jian-Dong Li, Milan Mrksich, Adegboyega K. Oyelere
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 24) pp:7543-7564
Publication Date(Web):15 December 2015
DOI:10.1016/j.bmc.2015.10.045
Inhibition of the enzymatic activity of histone deacetylase (HDAC) is a promising therapeutic strategy for cancer treatment and several distinct small molecule histone deacetylase inhibitors (HDACi) have been reported. We have previously identified a new class of non-peptide macrocyclic HDACi derived from 14- and 15-membered macrolide skeletons. In these HDACi, the macrocyclic ring is linked to the zinc chelating hydroxamate moiety through a para-substituted aryl-triazole cap group. To further delineate the depth of the SAR of this class of HDACi, we have synthesized series of analogous compounds and investigated the influence of various substitution patterns on their HDAC inhibitory, anti-proliferative and anti-inflammatory activities. We identified compounds 25b and 38f with robust anti-proliferative activities and compound 26f (IC50 47.2 nM) with superior anti-inflammatory (IC50 88 nM) activity relative to SAHA.
Co-reporter:Arren Z. Washington, Subhasish Tapadar, Alex George, Adegboyega K. Oyelere
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 16) pp:5198-5209
Publication Date(Web):15 August 2015
DOI:10.1016/j.bmc.2015.04.078
The ribosome is the primary protein synthesis machine in the cell and is a target for treatment of a variety of diseases including bacterial infection and cancer. The ribosomal peptide exit tunnel, the route of egress for the nascent peptide, is an inviting site for drug design. Toward a rational engagement of the nascent peptide components for the design of small molecule inhibitors of ribosome function, we designed and disclosed herein a set of N-10 indole functionalized azithromycin analogs. The indole moiety of these compounds is designed to mimic the translation stalling interaction of SecM W155 side-chain with the prokaryotic (Escherichia coli) ribosome A751 residue. Many of these N-10 functionalized compounds have enhanced translation inhibition activities against E. coli ribosome relative to azithromycin while a subset inhibited the growth of representative susceptible bacteria strains to about the same extent as azithromycin. Moreover, the inclusion of bovine serum in the bacterial growth media enhanced the anti-bacterial potency of the N-10 functionalized azithromycin analogs by as high as 10-fold.
Co-reporter:Arren Z. Washington, Derek B. Benicewicz, Joshua C. Canzoneri, Crystal E. Fagan, Sandra C. Mwakwari, Tatsuya Maehigashi, Christine M. Dunham, and Adegboyega K. Oyelere
ACS Chemical Biology 2014 Volume 9(Issue 11) pp:2621
Publication Date(Web):September 8, 2014
DOI:10.1021/cb5003224
Despite decades of research on the bacterial ribosome, the ribosomal exit tunnel is still poorly understood. Although it has been suggested that the exit tunnel is simply a convenient route of egress for the nascent chain, specific protein sequences serve to slow the rate of translation, suggesting some degree of interaction between the nascent peptide chain and the exit tunnel. To understand how the ribosome interacts with nascent peptide sequences, we synthesized and characterized a novel class of probe molecules. These peptide–macrolide (or “peptolide”) conjugates were designed to present unique peptide sequences to the exit tunnel. Biochemical and X-ray structural analyses of the interactions between these probes and the ribosome reveal interesting insights about the exit tunnel. Using translation inhibition and RNA structure probing assays, we find the exit tunnel has a relaxed preference for the directionality (N → C or C → N orientation) of the nascent peptides. Moreover, the X-ray crystal structure of one peptolide derived from a positively charged, reverse Nuclear Localization Sequence peptide, bound to the 70S bacterial ribosome, reveals that the macrolide ring of the peptolide binds in the same position as other macrolides. However, the peptide tail folds over the macrolide ring, oriented toward the peptidyl transferase center and interacting in a novel manner with 23S rRNA residue C2442 and His69 of ribosomal protein L4. These data suggest that these peptolides are viable probes for interrogating nascent peptide–exit tunnel interaction.
Co-reporter:Quaovi Sodji, Vishal Patil, Surendra Jain, James R. Kornacki, Milan Mrksich, Babu L. Tekwani, Adegboyega K. Oyelere
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 20) pp:4826-4830
Publication Date(Web):15 October 2014
DOI:10.1016/j.bmcl.2014.08.060
Histone deacetylase inhibitors (HDACi) pleiotropy is largely due to their nonselective inhibition of various cellular HDAC isoforms. Connecting inhibition of a specific isoform to biological responses and/or phenotypes is essential toward deconvoluting HDACi pleiotropy. The contribution of classes I and II HDACs to the antileishmanial activity of HDACi was investigated using the amastigote and promastigote forms of Leishmania donovani. We observed that the antileishmanial activities of HDACi are largely due to the inhibition of HDAC6-like activity. This observation could facilitate the development of HDACi as antileishmanial agents.Graphical abstract
Co-reporter:Vishal Patil ; Quaovi H. Sodji ; James R. Kornacki ; Milan Mrksich
Journal of Medicinal Chemistry 2013 Volume 56(Issue 9) pp:3492-3506
Publication Date(Web):April 2, 2013
DOI:10.1021/jm301769u
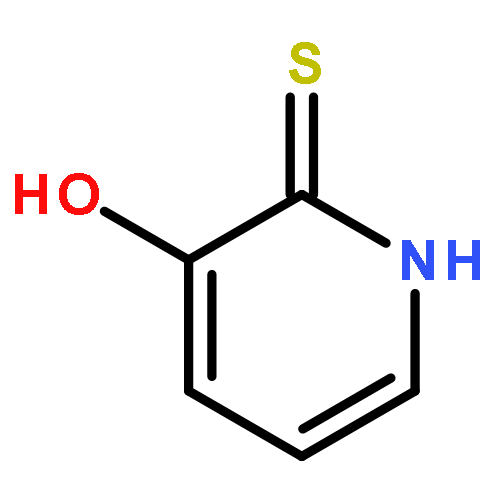
Small molecules bearing hydroxamic acid as the zinc binding group (ZBG) have been the most effective histone deacetylase inhibitors (HDACi) to date. However, concerns about the pharmacokinetic liabilities of the hydroxamic acid moiety have stimulated research efforts aimed at finding alternative nonhydroxamate ZBGs. We have identified 3-hydroxypyridin-2-thione (3-HPT) as a novel ZBG that is compatible with HDAC inhibition. 3-HPT inhibits HDAC 6 and HDAC 8 with an IC50 of 681 and 3675 nM, respectively. Remarkably, 3-HPT gives no inhibition of HDAC 1. Subsequent optimization led to several novel 3HPT-based HDACi that are selective for HDAC 6 and HDAC 8. Furthermore, a subset of these inhibitors induces apoptosis in various cancer cell lines.
Co-reporter:Berkley E. Gryder ; Michael K. Rood ; Kenyetta A. Johnson ; Vishal Patil ; Eric D. Raftery ; Li-Pan D. Yao ; Marcie Rice ; Bahareh Azizi ; Donald F. Doyle
Journal of Medicinal Chemistry 2013 Volume 56(Issue 14) pp:5782-5796
Publication Date(Web):June 20, 2013
DOI:10.1021/jm400467w
We describe a set of novel histone deacetylase inhibitors (HDACi) equipped with either an antagonist or an agonist of the estrogen receptor (ER) to confer selective activity against breast cancers. These bifunctional compounds potently inhibit HDAC at nanomolar concentrations and either agonize or antagonize ERα and ERβ. The ER antagonist activities of tamoxifen–HDACi conjugates (Tam-HDACi) are nearly identical to those of tamoxifen. Conversely, ethynyl-estradiol–HDACi conjugates (EED-HDACi) have attenuated ER agonist activities relative to the parent ethynyl-estradiol. In silico docking analysis provides structural basis for the trends of ER agonism/antagonism and ER subtype selectivity. Excitingly, lead Tam-HDACi conjugates show anticancer activity that is selectively more potent against MCF-7 (ERα positive breast cancer) compared to MDA-MB-231 (triple negative breast cancer), DU145 (prostate cancer), or Vero (noncancerous cell line). This dual-targeting approach illustrates the utility of designing small molecules with an emphasis on cell-type selectivity, not merely improved potency, working toward a higher therapeutic index at the earliest stages of drug development.
Co-reporter:Quaovi H. Sodji ; Vishal Patil ; James R. Kornacki ; Milan Mrksich
Journal of Medicinal Chemistry 2013 Volume 56(Issue 24) pp:9969-9981
Publication Date(Web):December 4, 2013
DOI:10.1021/jm401225q
We previously identified 3-hydroxypyridine-2-thione (3HPT) as a novel zinc binding group for histone deacetylase (HDAC) inhibition. Early structure–activity relationship (SAR) studies led to various small molecules possessing selective inhibitory activity against HDAC6 or HDAC8 but devoid of HDAC1 inhibition. To delineate further the depth of the SAR of 3HPT-derived HDAC inhibitors (HDACi), we have extended the SAR studies to include the linker region and the surface recognition group to optimize the HDAC inhibition. The current efforts resulted in the identification of two lead compounds, 10d and 14e, with potent HDAC6 and HDAC8 activities that are inactive against HDAC1. These new HDACi possess anticancer activities against various cancer cell lines including Jurkat J.γ1 for which SAHA and the previously disclosed 3HPT-derived HDACi were inactive.
Co-reporter:Berkley E. Gryder, Michelle J. Akbashev, Michael K. Rood, Eric D. Raftery, Warren M. Meyers, Paulette Dillard, Shafiq Khan, and Adegboyega K. Oyelere
ACS Chemical Biology 2013 Volume 8(Issue 11) pp:2550
Publication Date(Web):September 4, 2013
DOI:10.1021/cb400542w
Diverse cellular processes relevant to cancer progression are regulated by the acetylation status of proteins. Among such processes is chromatin remodeling via histone proteins, controlled by opposing histone deacetylase (HDAC) and histone acetyltransferase (HAT) enzymes. Histone deacetylase inhibitors (HDACi) show great promise in preclinical cancer models, but clinical trials treating solid tumors have failed to improve patient survival. This is due in part to an inability of HDACi to effectively accumulate in cancerous cells. To address this problem we designed HDACi with secondary pharmacophores to facilitate selective accumulation in malignant cells. We present the first example of HDACi compounds targeted to prostate tumors by equipping them with the additional ability to bind the androgen receptor (AR) with nonsteroidal antiandrogen moieties. Leads among these new dual-acting molecules bind to the AR and halt AR transcriptional activity at lower concentrations than clinical antiandrogens. They inhibit key isoforms of HDAC with low nanomolar potency. Fluorescent microscopy reveals varying degrees of AR nuclear localization in response to these compounds that correlates with their HDAC activity. These biological properties translate into potent anticancer activity against hormone-dependent (AR+) LNCaP and to a lesser extent against hormone-independent (AR−) DU145 prostate cancer, while having greatly reduced toxicity in noncancerous cells. This illustrates that engaging multiple biological targets with a single chemical probe can achieve both potent and cell-type-selective responses.
Co-reporter:William Guerrant, Vishal Patil, Joshua C. Canzoneri, Li-Pan Yao, Rebecca Hood, Adegboyega K. Oyelere
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 11) pp:3283-3287
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmcl.2013.03.108
Current chemotherapy regimens are comprised mostly of single-target drugs which are often plagued by toxic side effects and resistance development. A pharmacological strategy for circumventing these drawbacks could involve designing multivalent ligands that can modulate multiple targets while avoiding the toxicity of a single-targeted agent. Two attractive targets, histone deacetylase (HDAC) and topoisomerase I (Topo I), are cellular modulators that can broadly arrest cancer proliferation through a range of downstream effects. Both are clinically validated targets with multiple inhibitors in therapeutic use. We describe herein the design and synthesis of dual-acting histone deacetylase–topoisomerase I inhibitors. We also show that these dual-acting agents retain activity against HDAC and Topo I, and potently arrest cancer proliferation.
Co-reporter:William Guerrant ; Vishal Patil ; Joshua C. Canzoneri
Journal of Medicinal Chemistry 2012 Volume 55(Issue 4) pp:1465-1477
Publication Date(Web):January 19, 2012
DOI:10.1021/jm200799p
Strategies to ameliorate the flaws of current chemotherapeutic agents, while maintaining potent anticancer activity, are of particular interest. Agents which can modulate multiple targets may have superior utility and fewer side effects than current single-target drugs. To explore the prospect in cancer therapy of a bivalent agent that combines two complementary chemo-active groups within a single molecular architecture, we have synthesized dual-acting histone deacetylase and topoisomerase II inhibitors. These dual-acting agents are derived from suberoylanilide hydroxamic acid (SAHA) and anthracycline daunorubicin, prototypical histone deacetylase (HDAC) and topoisomerase II (Topo II) inhibitors, respectively. We report herein that these agents present the signatures of inhibition of HDAC and Topo II in both cell-free and whole-cell assays. Moreover, these agents potently inhibit the proliferation of representative cancer cell lines.
Co-reporter:Erik C. Dreaden, Berkley E. Gryder, Lauren A. Austin, Brice A. Tene Defo, Steven C. Hayden, Min Pi, L. Darryl Quarles, Adegboyega K. Oyelere, and Mostafa A. El-Sayed
Bioconjugate Chemistry 2012 Volume 23(Issue 8) pp:1507
Publication Date(Web):July 9, 2012
DOI:10.1021/bc300158k
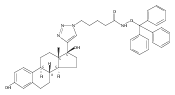
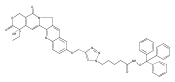
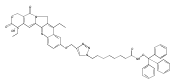
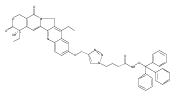
Prostate cancer is the most commonly diagnosed cancer among men in the developed countries.(1) One in six males in the U.S.(2) and one in nine males in the U.K.(3) will develop the disease at some point during their lifetime. Despite advances in prostate cancer screening, more than a quarter of a million men die from the disease every year(1) due primarily to treatment-resistance and metastasis. Colloidal nanotechnologies can provide tremendous enhancements to existing targeting/treatment strategies for prostate cancer to which malignant cells are less sensitive. Here, we show that antiandrogen gold nanoparticles—multivalent analogues of antiandrogens currently used in clinical therapy for prostate cancer—selectively engage two distinct receptors, androgen receptor (AR), a target for the treatment of prostate cancer, as well as a novel G-protein coupled receptor, GPRC6A, that is also upregulated in prostate cancer. These nanoparticles selectively accumulated in hormone-insensitive and chemotherapy-resistant prostate cancer cells, bound androgen receptor with multivalent affinity, and exhibited greatly enhanced drug potency versus monovalent antiandrogens currently in clinical use. Further, antiandrogen gold nanoparticles selectively stimulated GPRC6A with multivalent affinity, demonstrating that the delivery of nanoscale antiandrogens can also be facilitated by the transmembrane receptor in order to realize increasingly selective, increasingly potent therapy for treatment-resistant prostate cancers.
Co-reporter:Berkley E. Gryder, Will Guerrant, Chin Ho Chen and Adegboyega K. Oyelere
MedChemComm 2011 vol. 2(Issue 11) pp:1083-1086
Publication Date(Web):16 Sep 2011
DOI:10.1039/C1MD00208B
Oxathiazole-2-one is a new candidate for proteasome inhibition which has not been widely explored. We describe herein the synthesis and characterization of a new oxathiazole-2-one derived from the dipeptide backbone of Bortezomib. We found that this new oxathiazole-2-one compound 1 is modestly active against the human 20S proteasome, but surprisingly has no significant activity against the M. tuberculosis proteasome. Additionally, the compound has improved aqueous stability compared to previously reported oxathiazole-2-one compounds. Molecular docking analyses provided information on the structural basis of the observed disparity between the human and mycobacterium proteasomes inhibitory activity of compound 1.
Co-reporter:Sandra C. Mwakwari ; William Guerrant ; Vishal Patil ; Shabana I. Khan ; Babu L. Tekwani ; Zachary A. Gurard-Levin ; Milan Mrksich
Journal of Medicinal Chemistry 2010 Volume 53(Issue 16) pp:6100-6111
Publication Date(Web):July 29, 2010
DOI:10.1021/jm100507q
Inhibition of histone deacetylase (HDAC) function is a validated therapeutic strategy for cancer treatment. Of the several structurally distinct small molecule histone deacetylase inhibitors (HDACi) reported, macrocyclic depsipeptides possess the most complex cap groups and have demonstrated excellent HDAC inhibition potency and isoform selectivity. Unfortunately, the development of macrocyclic depsipeptides has been hampered in part because of development problems characteristic of large peptides and the complex reaction schemes required for their synthesis. Herein we report that tricyclic ketolide TE-802 is an excellent mimetic for the peptide backbone of macrocyclic HDACi. Compounds derived from this template are particularly selective against HDACs 1 and 2 with nanomolar inhibitory activity. Interrogation of the association between a subset of these compounds and key HDAC isoforms, using AutoDock, enables a molecular description of the interaction between the HDAC enzyme’s outer rim and the inhibitors’ macrocyclic cap group that are responsible for compound affinity and presumably isoform selectivity.
Co-reporter:Vishal Patil, William Guerrant, Po C. Chen, Berkley Gryder, Derek B. Benicewicz, Shabana I. Khan, Babu L. Tekwani, Adegboyega K. Oyelere
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 1) pp:415-425
Publication Date(Web):1 January 2010
DOI:10.1016/j.bmc.2009.10.042
Histone deacetylase inhibitors (HDACi) are endowed with plethora of biological functions including anti-proliferative, anti-inflammatory, anti-parasitic, and cognition-enhancing activities. Parsing the structure–activity relationship (SAR) for each disease condition is vital for long-term therapeutic applications of HDACi. We report in the present study specific cap group substitution patterns and spacer-group chain lengths that enhance the antimalarial and antileishmanial activity of aryltriazolylhydroxamates-based HDACi. We identified many compounds that are several folds selectively cytotoxic to the plasmodium parasites compared to standard HDACi. Also, a few of these compounds have antileishmanial activity that rivals that of miltefosine, the only currently available oral agent against visceral leishmaniasis. The anti-parasite properties of several of these compounds tracked well with their anti-HDAC activities. The results presented here provide further evidence on the suitability of HDAC inhibition as a viable therapeutic option to curb infections caused by apicomplexan protozoans and trypanosomatids.
Co-reporter:William Guerrant;SraC. Mwakwari;PoC. Chen;ShabanaI. Khan;BabuL. Tekwani Dr.;AdegboyegaK. Oyelere
ChemMedChem 2010 Volume 5( Issue 8) pp:1232-1235
Publication Date(Web):
DOI:10.1002/cmdc.201000087
Co-reporter:Adegboyega K. Oyelere ; Po C. Chen ; William Guerrant ; Sandra C. Mwakwari ; Rebecca Hood ; Yunzhe Zhang ;Yuhong Fan
Journal of Medicinal Chemistry 2009 Volume 52(Issue 2) pp:456-468
Publication Date(Web):December 18, 2008
DOI:10.1021/jm801128g
Inhibition of histone deacetylase inhibitors (HDACi) hold great promise in cancer therapy because of their demonstrated ability to arrest proliferation of nearly all transformed cell types. Of the several structurally distinct small molecule HDACi reported, macrocyclic depsipeptides have the most complex recognition cap-group moieties and present an excellent opportunity for the modulation of the biological activities of HDACi. Unfortunately, the structure−activity relationship (SAR) studies for this class of compounds have been impaired largely because most macrocyclic HDACi known to date comprise complex peptide macrocycles. In addition to retaining the pharmacologically disadvantaged peptidyl backbone, they offer only limited opportunity for side chain modifications. Here, we report the discovery of a new class of macrocyclic HDACi based on the macrolide antibiotics skeletons. SAR studies revealed that these compounds displayed both linker-length and macrolide-type dependent HDAC inhibition activities with IC50 in the low nanomolar range. In addition, these non-peptide macrocyclic HDACi are more selective against HDACs 1 and 2 relative to HDAC 8, another class I HDAC isoform, and hence have subclass HDAC isoform selectivity.
Co-reporter:Erik C. Dreaden, Sandra C. Mwakwari, Quaovi H. Sodji, Adegboyega K. Oyelere and Mostafa A. El-Sayed
Bioconjugate Chemistry 2009 Volume 20(Issue 12) pp:2247
Publication Date(Web):November 17, 2009
DOI:10.1021/bc9002212
The breast cancer treatment drug tamoxifen has been widely administered for more than three decades. This small molecule competes with 17β-estradiol for binding to estrogen receptor, a hormone receptor upregulated in a majority of breast cancers, subsequently initiating programmed cell death. We have synthesized a thiol-PEGylated tamoxifen derivative that can be used to selectively target and deliver plasmonic gold nanoparticles to estrogen receptor positive breast cancer cells with up to 2.7-fold enhanced drug potency in vitro. Optical microscopy/spectroscopy, time-dependent dose−response data, and estrogen competition studies indicate that augmented activity is due to increased rates of intracellular tamoxifen transport by nanoparticle endocytosis, rather than by passive diffusion of the free drug. Both ligand- and receptor-dependent intracellular delivery of gold nanoparticles suggest that plasma membrane localized estrogen receptor alpha may facilitate selective uptake and retention of this and other therapeutic nanoparticle conjugates. Combined targeting selectivity and enhanced potency provides opportunities for both multimodal endocrine treatment strategies and adjunctive laser photothermal therapy.
Co-reporter:Joshua C. Canzoneri, Po C. Chen, Adegboyega K. Oyelere
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 23) pp:6588-6590
Publication Date(Web):1 December 2009
DOI:10.1016/j.bmcl.2009.10.028
We describe herein the synthesis and characterization of a new class of histone deacetylase (HDAC) inhibitors derived from conjugation of a suberoylanilide hydroxamic acid-like aliphatic-hydroxamate pharmacophore to a nuclear localization signal peptide. We found that these conjugates inhibited the histone deacetylase activities of HDACs 1, 2, 6, and 8 in a manner similar to suberoylanilide hydroxamic acid (SAHA). Notably, compound 7b showed a threefold improvement in HDAC 1/2 inhibition, a threefold increase in HDAC 6 selectivity and a twofold increase in HDAC 8 selectivity when compared to SAHA.
Co-reporter:Idris Raji, Kabir Ahluwalia, Adegboyega K. Oyelere
Bioorganic & Medicinal Chemistry Letters (15 February 2017) Volume 27(Issue 4) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.bmcl.2017.01.044
The clinical validation of histone deacetylase inhibition as a cancer therapeutic modality has stimulated interest in the development of new generation of potent and tumor selective histone deacetylase inhibitors (HDACi). With the goal of selective delivery of the HDACi to melanoma cells, we incorporated the benzamide, a high affinity melanin-binding template, into the design of HDACi to generate a new series of compounds 10a-b and 11a-b which display high potency towards HDAC1 and HDAC6. However, these compounds have attenuated antiproliferative activities relative to the untargeted HDACi. An alternative strategy furnished compound 14, a prodrug bearing the benzamide template linked via a labile bond to a hydroxamate-based HDACi. This pro-drug compound showed promising antiproliferative activity and warrant further study.
Co-reporter:Idris Raji, Fatima Yadudu, Emily Janeira, Shaghayegh Fathi, Lindsey Szymczak, James Richard Kornacki, Kensei Komatsu, Jian-Dong Li, Milan Mrksich, Adegboyega K. Oyelere
Bioorganic & Medicinal Chemistry (1 February 2017) Volume 25(Issue 3) pp:
Publication Date(Web):1 February 2017
DOI:10.1016/j.bmc.2016.12.032
We herein disclose a series of compounds with potent inhibitory activities towards histone deacetylases (HDAC) and cyclooxygenases (COX). These compounds potently inhibited the growth of cancer cell lines consistent with their anti-COX and anti-HDAC activities. While compound 2b showed comparable level of COX-2 selectivity as celecoxib, compound 11b outperformed indomethacin in terms of selectivity towards COX-2 relative to COX-1. An important observation with our lead compounds (2b, 8, 11b, and 17b) is their enhanced cytotoxicity towards androgen dependent prostate cancer cell line (LNCaP) relative to androgen independent prostate cancer cell line (DU-145). Interestingly, compounds 2b and 17b arrested the cell cycle progression of LNCaP in the S-phase, while compound 8 showed a G0/G1 arrest, similar to SAHA. Relative to SAHA, these compounds displayed tumor-selective cytotoxicity as they have low anti-proliferative activity towards healthy cells (VERO); an attribute that makes them attractive candidates for drug development.

![Benzenemethanamine, N-[2-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethyl]-4-ethynyl-N-methyl-](/data/chemimg/178200/1445868-72-3.png)
![Benzenemethanamine, N-[2-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethyl]-4-ethynyl-N-methyl-](/data/chemimg/178200/1445868-72-3_b.png)
![Octanediamide, N1-[4-[[[2-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethyl]methylamino]methyl]phenyl]-N8-hydroxy-](/data/chemimg/178200/1445868-71-2.png)
![Octanediamide, N1-[4-[[[2-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethyl]methylamino]methyl]phenyl]-N8-hydroxy-](/data/chemimg/178200/1445868-71-2_b.png)
![Octanoic acid, 8-[[4-[[[2-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethyl]methylamino]methyl]phenyl]amino]-8-oxo-, methyl ester](/data/chemimg/178200/1445868-70-1.png)
![Octanoic acid, 8-[[4-[[[2-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethyl]methylamino]methyl]phenyl]amino]-8-oxo-, methyl ester](/data/chemimg/178200/1445868-70-1_b.png)










































































![Carbamic acid, [2-nitro-4-(2-thienyl)phenyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/335300/335254-95-0.png)
![Carbamic acid, [2-nitro-4-(2-thienyl)phenyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/335300/335254-95-0_b.png)






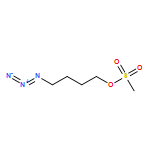
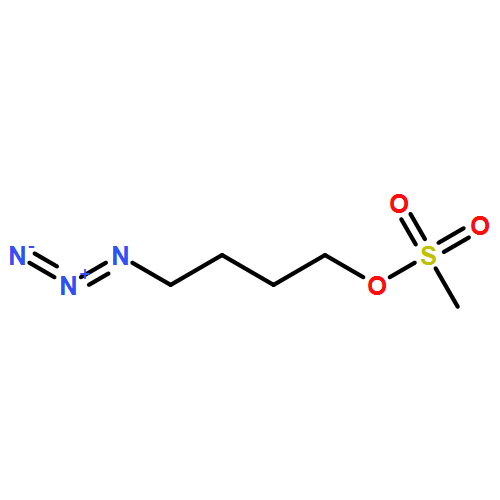
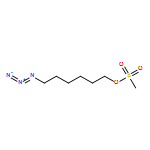

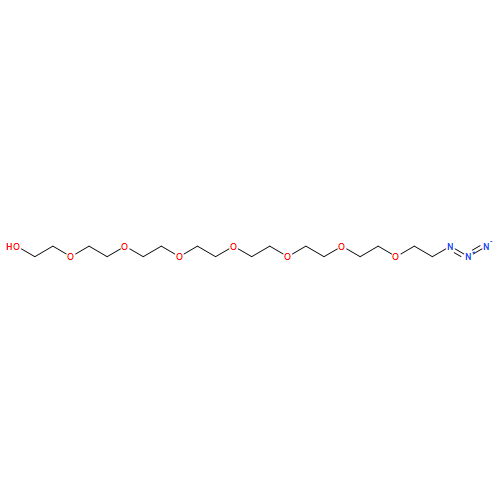
![4-[5-(4-methylphenyl)-3-trifluoromethyl-1H-pyrazol-1-yl]benzenesulfonyl chloride](http://img.cochemist.com/ccimg/190800/190711-56-9.png)
![4-[5-(4-methylphenyl)-3-trifluoromethyl-1H-pyrazol-1-yl]benzenesulfonyl chloride](http://img.cochemist.com/ccimg/190800/190711-56-9_b.png)




![1H-Pyrazole-3-carboxylic acid,1-[4-(aminosulfonyl)phenyl]-5-(4-methylphenyl)-, ethyl ester](http://img.cochemist.com/ccimg/170600/170569-23-0.png)
![1H-Pyrazole-3-carboxylic acid,1-[4-(aminosulfonyl)phenyl]-5-(4-methylphenyl)-, ethyl ester](http://img.cochemist.com/ccimg/170600/170569-23-0_b.png)

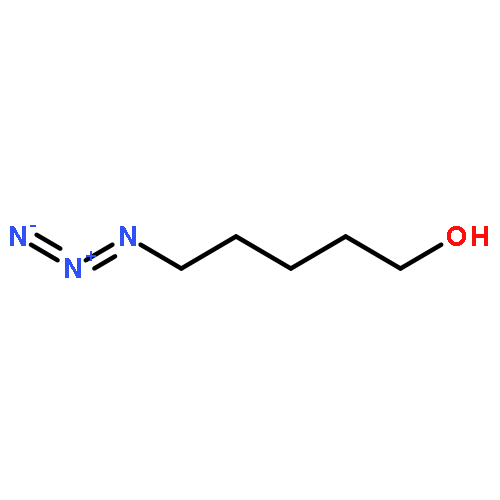

















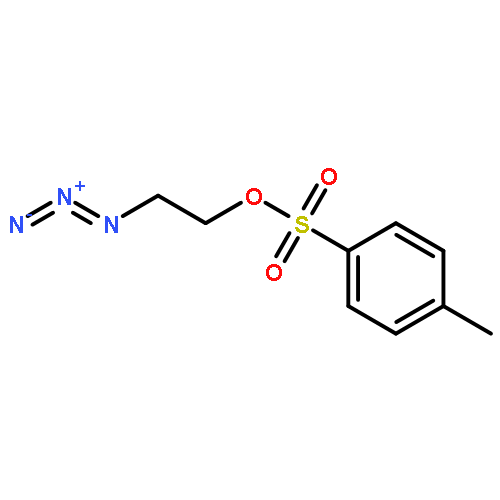

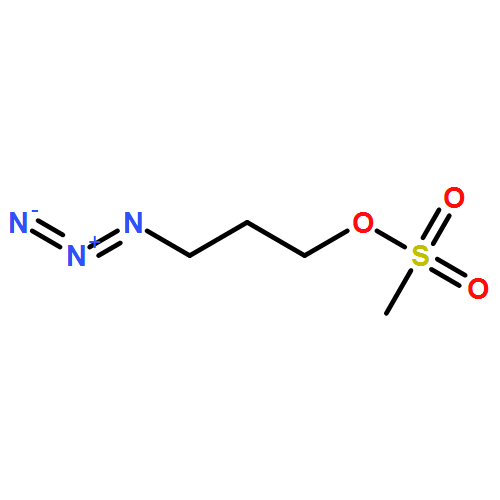








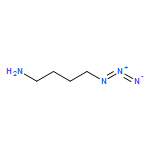
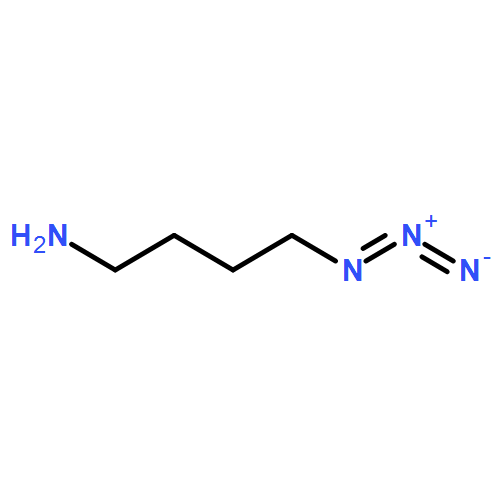
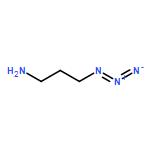
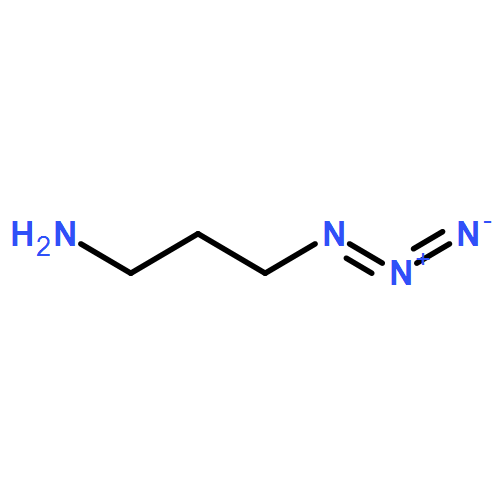


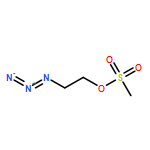
























![Octanoic acid, 8-[[4-[[(methylsulfonyl)oxy]methyl]phenyl]amino]-8-oxo-, methyl ester](/data/chemimg/178200/1093857-04-5.png)
![Octanoic acid, 8-[[4-[[(methylsulfonyl)oxy]methyl]phenyl]amino]-8-oxo-, methyl ester](/data/chemimg/178200/1093857-04-5_b.png)











![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7.png)
![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7_b.png)