Co-reporter:Dongwei Kang, Xiao Ding, Gaochan Wu, Zhipeng Huo, Zhongxia Zhou, Tong Zhao, Da Feng, Zhao Wang, Ye Tian, Dirk Daelemans, Erik De Clercq, Christophe Pannecouque, Peng Zhan, and Xinyong Liu
ACS Medicinal Chemistry Letters November 9, 2017 Volume 8(Issue 11) pp:1188-1188
Publication Date(Web):October 19, 2017
DOI:10.1021/acsmedchemlett.7b00361
Our previous studies led us to conclude that thiophene[3,2-d]pyrimidine is a promising scaffold for diarylpyrimidine (DAPY)-type anti-HIV agents with potent activity against resistance-associated human immunodeficiency virus (HIV) variants (J. Med. Chem. 2016, 59, 7991–8007; J. Med. Chem. 2017, 60, 4424–4443). In the present study, we designed and synthesized a series of thiophenepyrimidine derivatives with various substituents in the right wing region of the structure with the aim of developing new interactions with the tolerant region I of the binding pocket of the HIV-1 non-nucleoside reverse transcriptase (NNRTI), and we evaluated their activity against a panel of mutant HIV-1 strains. All the derivatives exhibited moderate to excellent potency against wild-type (WT) HIV-1 in MT-4 cells. Among them, sulfonamide compounds 9b and 9d were single-figure-nanomolar inhibitors with EC50 values of 9.2 and 7.1 nM, respectively. Indeed, 9a and 9d were effective against the whole viral panel except RES056. Notably, both compounds showed potent antiviral activity against K103N (EC50 = 0.032 and 0.070 μM) and E138K (EC50 = 0.035 and 0.045 μM, respectively). Furthermore, 9a and 9d exhibited high affinity for WT HIV-1 RT (IC50 = 1.041 and 1.138 μM, respectively) and acted as classical NNRT inhibitors (NNRTIs). These results are expected to be helpful in the design of thiophenepyrimidine-based NNRTIs with more potent activity against HIV strains with RT mutations.Keywords: drug design; HIV-1; NNRTIs; thiophene[3,2-d]pyrimidine; tolerant region I;
Co-reporter:Dongwei Kang, Zengjun Fang, Boshi Huang, Xueyi Lu, Heng Zhang, Haoran Xu, Zhipeng Huo, Zhongxia Zhou, Zhao Yu, Qing Meng, Gaochan Wu, Xiao Ding, Ye Tian, Dirk Daelemans, Erik De Clercq, Christophe Pannecouque, Peng Zhan, and Xinyong Liu
Journal of Medicinal Chemistry May 25, 2017 Volume 60(Issue 10) pp:4424-4424
Publication Date(Web):May 8, 2017
DOI:10.1021/acs.jmedchem.7b00332
This work follows on from our initial discovery of a series of piperidine-substituted thiophene[3,2-d]pyrimidine HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTI) ( J. Med. Chem. 2016, 59, 7991−8007). In the present study, we designed, synthesized, and biologically tested several series of new derivatives in order to investigate previously unexplored chemical space. Some of the synthesized compounds displayed single-digit nanomolar anti-HIV potencies against wild-type (WT) virus and a panel of NNRTI-resistant mutant viruses in MT-4 cells. Compound 25a was exceptionally potent against the whole viral panel, affording 3-4-fold enhancement of in vitro antiviral potency against WT, L100I, K103N, Y181C, Y188L, E138K, and K103N+Y181C and 10-fold enhancement against F227L+V106A relative to the reference drug etravirine (ETV) in the same cellular assay. The structure–activity relationships, pharmacokinetics, acute toxicity, and cardiotoxicity were also examined. Overall, the results indicate that 25a is a promising new drug candidate for treatment of HIV-1 infection.
Co-reporter:Han Ju, Jian Zhang, Boshi Huang, Dongwei Kang, Bing Huang, Xinyong Liu, and Peng Zhan
Journal of Medicinal Chemistry May 11, 2017 Volume 60(Issue 9) pp:3533-3533
Publication Date(Web):January 24, 2017
DOI:10.1021/acs.jmedchem.6b01227
Influenza virus (IFV) causes periodic global influenza pandemics, resulting in substantial socioeconomic loss and burden on medical facilities. Yearly variation in the effectiveness of vaccines, slow responsiveness to vaccination in cases of pandemic IFV, and emerging resistance to available drugs highlight the need to develop additional small-molecular inhibitors that act on IFV proteins. One promising target is polymerase acidic (PA) endonuclease, which is a bridged dinuclear metalloenzyme that plays a crucial role in initiating IFV replication. During the past decade, intensive efforts have been made to develop small-molecular inhibitors of this endonuclease as candidate agents for treatment of IFV infection. Here, we review the current status of development of PA endonuclease inhibitors and we discuss the applicability of newer medicinal-chemistry strategies for the discovery more potent, selective, and safer inhibitors.
Co-reporter:Zhaoqiang Liu, Ye Tian, Jinghan Liu, Boshi Huang, Dongwei Kang, Erik De Clercq, Dirk Daelemans, Christophe Pannecouque, Peng Zhan, Xinyong Liu
European Journal of Medicinal Chemistry 2017 Volume 140(Volume 140) pp:
Publication Date(Web):10 November 2017
DOI:10.1016/j.ejmech.2017.07.012
•Twenty-one 3,5-diaryl-pyridine derivatives were designed, synthesized and evaluated for their anti-HIV activities.•5b2 (EC50 = 0.04 μM, SI = 3963) was the most potent inhibitor.•The 2-NH2 pyridine derivatives showed better anti-HIV-1 activity than the corresponding 2-NO2 counterparts.As a continuation of our efforts to discover and develop “me-better” drugs of DAPYs, novel diarylpyridine derivatives were designed, synthesized and evaluated for their anti-HIV activities in MT-4 cells. The majority of these compounds showed high activity against wild-type HIV-1 strain (IIIB) with EC50 values in the range of 0.04–4.41 μM. Among them, compound 5b2 (EC50 = 0.04 μM, SI = 3963) was the most potent. This compound showed anti-HIV-1IIIB activity superior than of Nevirapine but still inferior than of Etravirine. Selected compounds were also evaluated for the activity against reverse transcriptase (RT), and most of the compounds exhibited submicromolar IC50 values indicating they are specific RT inhibitors. Preliminary structure-activity relationships and modeling studies of these new analogues provide valuable avenues for future molecular optimization.Download high-res image (361KB)Download full-size image
Co-reporter:Heng Zhang, Ye Tian, Dongwei Kang, Zhipeng Huo, Zhongxia Zhou, Huiqing Liu, Erik De Clercq, Christophe Pannecouque, Peng Zhan, Xinyong Liu
European Journal of Medicinal Chemistry 2017 Volume 130(Volume 130) pp:
Publication Date(Web):21 April 2017
DOI:10.1016/j.ejmech.2017.02.047
•Novel uracil-bearing DAPYs derivatives were identified.•16d exhibited best activity with EC50 values of 5.6 nM (wt) and 34.2 nM (E138K).•LogP and water solubility of representative compounds were measured.•Preliminary SARs and molecular simulation of these new analogues were detailed.A novel series of uracil-bearing DAPYs derivatives were designed and synthesized via structure-based molecular hybridization to discover compounds with improved anti-resistance profiles. Anti-HIV activity of the designed compounds was tested in MT-4 cell cultures. The most promising compound 16d showed excellent activity with EC50 value of 5.6 nM against wide-type HIV-1 and low cytotoxicity (SI > 50000). Activity against the clinic prevalent mutant strains was also tested, suggesting that 16d was sensitive to E138K (EC50 = 34.2 nM). Primary drug-like properties, such as water solubility and logP, were evaluated by experiment or calculation, which indicated that introducing an uracil can improve solubility. The molecular modeling accompanied with the preliminary SAR correlations paved the way for the next round of rational design of potent anti-HIV agents.By structure-based molecular hybridization, a new HIV-1 NNRTI 16d was discovered with excellent activity and high selectivity.Download high-res image (164KB)Download full-size image
Co-reporter:Xiao Li, Ping Gao, Boshi Huang, Zhongxia Zhou, Zhao Yu, Zheng Yuan, Huiqing Liu, Christophe Pannecouque, Dirk Daelemans, Erik De Clercq, Peng Zhan, Xinyong Liu
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.10.009
•Novel IASs bearing N-substituted piperidine at indole-2-carboxamide were designed.•New IASs have EC50 values ranging from 0.62 μM to 0.006 μM against WT HIV-1.•Compounds 8 (EC50 = 6 nM) and 18 (EC50 = 9 nM) were the most active derivatives.•Compounds 8 and 18 also displayed outstanding potency against some HIV-1 mutants.•SARs and molecular modeling studies were discussed in detail.To further explore the chemical space around the entrance channel of HIV-1 reverse transcriptase (RT), a series of novel indolylarylsulfones (IASs) bearing N-substituted piperidine at indole-2-carboxamide were identified as potent HIV NNRTIs by structure-guided scaffold morphing and fragment rearrangement. All the IASs exhibited moderate to excellent potency against wild-type HIV-1 with EC50 values ranging from 0.62 μM to 0.006 μM 8 (EC50 = 6 nM) and 18 (EC50 = 9 nM) were identified as the most potent compounds, which were more active than NVP and DLV, and reached the same order of EFV and ETV. Furthermore, most compounds maintained high activity agaist various single HIV-1 mutants (L100I, K103N, E138K, Y181C) as well as one double mutant (F227L/V106A) with EC50 values in low-micromolar to double-digit nanomolar concentration ranges. Especially, 8 displayed outstanding potency against L100I (EC50 = 17 nM with a 2.8-fold resistance ratio) and 18 was relatively more potent to E138K mutant (EC50 = 43 nM with a 4.7-fold resistance ratio). Preliminary SARs and molecular modeling studies were also discussed in detail, which may provide valuable insights for further optimization.A series of novel indolylarylsulfones (IASs) bearing N-substituted piperidine at indole-2-carboxamide were identified as potent HIV NNRTIs by structure-guided scaffold morphing and fragment rearrangement.Download high-res image (190KB)Download full-size image
Co-reporter:Boshi Huang, Xueshun Wang, Xinhao Liu, Zihui Chen, Wanzhuo Li, Songkai Sun, Huiqing Liu, Dirk Daelemans, Erik De Clercq, Christophe Pannecouque, Peng Zhan, Xinyong Liu
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 16(Issue 16) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.bmc.2017.06.022
Crystallographic overlap studies and pharmacophoric analysis indicated that diarylpyrimidine (DAPY)-based HIV-1 NNRTIs showed a similar binding mode and pharmacophoric features as indolylarylsulfones (IASs), another class of potent NNRTIs. Thus, a novel series of DAPY-IAS hybrid derivatives were identified as newer NNRTIs using structure-based molecular hybridization. Some target compounds exhibited moderate activities against HIV-1 IIIB strain, among which the two most potent inhibitors possessed EC50 values of 1.48 μM and 1.61 μM, respectively. They were much potent than the reference drug ddI (EC50 = 76.0 μM) and comparable to 3TC (EC50 = 2.54 μM). Compound 7a also exhibited the favorable selectivity index (SI = 80). Preliminary structure-activity relationships (SARs), structure-cytotoxicity relationships, molecular modeling studies, and in silico calculation of physicochemical properties of these new inhibitors were also discussed.A novel series of DAPY-IAS hybrid derivatives were identified as newer HIV-1 NNRTIs using structure-based molecular hybridization.Download high-res image (159KB)Download full-size image
Co-reporter:Tao Zhang, Min Zhai, Jianbo Ji, Jian Zhang, Ye Tian, Xinyong Liu
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 11(Issue 11) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.bmcl.2017.04.028
Ebola virus is one of the most threatening pathogens with the mortality rate as high as 90% in the world. There are no licensed therapeutic drugs or preventive vaccines for Ebola hemorrhagic fever up to date. Favipiravir, a novel antiviral drug which was mainly used for the treatment of influenza, now has been demonstrated to have a curative effect in treating Ebola virus infection. In this review, we present an overview of recent progress on the treatment of Ebola virus disease with Favipiravir and describe its possible mechanism. Moreover, we give a brief summary of other related treatment strategies against Ebola.Download high-res image (56KB)Download full-size image
Co-reporter:Ping Gao, Lingzi Zhang, Lin Sun, Tianguang Huang, Jing Tan, Jian Zhang, Zhongxia Zhou, Tong Zhao, Luis Menéndez-Arias, Christophe Pannecouque, Erik De Clercq, Peng Zhan, Xinyong Liu
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 20(Issue 20) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.bmc.2017.09.006
A small library containing 3-hydroxyquinazoline-2,4(1H,3H)-dione and 1-hydroxypyrido[2,3-d]pyrimidin-2(1H)-one scaffolds was obtained via the copper(I)-catalyzed azidealkyne cycloaddition (CuAAC) reaction and evaluated for their anti-HIV activity in MT-4 cells. Among the synthesized compounds, several 1-hydroxypyrido[2,3-d]pyrimidin-2(1H)-one derivatives showed remarkable anti-HIV potency with EC50 values ranging from 0.92 to 26.85 µM. The most active one, IIA-2, also showed remarkable and selective potency against HIV type 1 integrase (IN). To the best of our knowledge, this is the first report showing that 1-hydroxypyrido[2,3-d]pyrimidin-2(1H)-ones are selective HIV IN inhibitors. Preliminary structure-activity relationship (SAR) studies suggested that the divalent metal ion chelators and the nature and position of substituents around the core are important for antiviral potency. Molecular modeling has been used to predict the binding site of the pyrido[2,3-d]pyrimidin-2(1H)-one core in HIV type 1 IN and suggestions are made for improvement of its inhibitory activity.
Co-reporter:Haiyong Jia, Yang Song, Ji Yu, Peng Zhan, Diwakar Rai, Xiaohong Liang, Chunhong Ma, Xinyong Liu
European Journal of Medicinal Chemistry 2017 Volume 136(Volume 136) pp:
Publication Date(Web):18 August 2017
DOI:10.1016/j.ejmech.2017.04.048
•26 pyridone derivatives were prepared.•Novel pyridone derivatives were identified as non-nucleoside HBV inhibitors.•Compound 8u suppressed the replication of HBV DNA with IC50 value of 3.4 μM.•Preliminary SARs of these new derivatives were detailed.In continuation of our efforts toward the discovery of potent non-nucleoside hepatitis B virus (HBV) inhibitors with novel structures, we have employed bioisosterism and hybrid pharmacophore-based strategy to explore the chemically diverse space of bioactive compounds. Cytotoxicity, anti-HBV antigen secretion activities and anti-HBV DNA replication activity were assayed with cell counting kit-8 (CCK-8), enzyme linked immunosorbent assay (ELISA) and a real-time PCR, respectively. Some of the new compounds were able to inhibit the replication of HBV DNA activity in the low micromolar range. In particular, compound 8u displayed the most potent activity against the replication of HBV DNA with IC50 value of 3.4 μM. The preliminary structure-activity relationship (SAR) of these new compounds was investigated, which may help designing more potent molecules.A series of novel pyridone derivatives were identified as non-nucleoside HBV inhibitors via bioisosterism and hybrid pharmacophore-strategy.Download high-res image (159KB)Download full-size image
Co-reporter:Dongwei Kang, Zengjun Fang, Zhenyu Li, Boshi Huang, Heng Zhang, Xueyi Lu, Haoran Xu, Zhongxia Zhou, Xiao Ding, Dirk Daelemans, Erik De Clercq, Christophe Pannecouque, Peng Zhan, and Xinyong Liu
Journal of Medicinal Chemistry 2016 Volume 59(Issue 17) pp:7991-8007
Publication Date(Web):August 19, 2016
DOI:10.1021/acs.jmedchem.6b00738
We designed and synthesized a series of human immunodeficiency virus type 1 (HIV-1) non-nucleoside reverse transcriptase inhibitors (NNRTIs) with a piperidine-substituted thiophene[3,2-d]pyrimidine scaffold, employing a strategy of structure-based molecular hybridization and substituent decorating. Most of the synthesized compounds exhibited broad-spectrum activity with low (single-digit) nanomolar EC50 values toward a panel of wild-type (WT), single-mutant, and double-mutant HIV-1 strains. Compound 27 was the most potent; compared with ETV, its antiviral efficacy was 3-fold greater against WT, 5–7-fold greater against Y181C, Y188L, E138K, and F227L+V106A, and nearly equipotent against L100I and K103N, though somewhat weaker against K103N+Y181C. Importantly, 27 has lower cytotoxicity (CC50 > 227 μM) and a huge selectivity index (SI) value (ratio of CC50/EC50) of >159101. 27 also showed favorable, drug-like pharmacokinetic and safety properties in rats in vivo. Molecular docking studies and the structure–activity relationships provide important clues for further molecular elaboration.
Co-reporter:Peng Zhan; Christophe Pannecouque; Erik De Clercq
Journal of Medicinal Chemistry 2016 Volume 59(Issue 7) pp:2849-2878
Publication Date(Web):October 28, 2015
DOI:10.1021/acs.jmedchem.5b00497
The early effectiveness of combinatorial antiretroviral therapy (cART) in the treatment of HIV infection has been compromised to some extent by rapid development of multidrug-resistant HIV strains, poor bioavailability, and cumulative toxicities, and so there is a need for alternative strategies of antiretroviral drug discovery and additional therapeutic agents with novel action modes or targets. From this perspective, we first review current strategies of antiretroviral drug discovery and optimization, with the aid of selected examples from the recent literature. We highlight the development of phosphate ester-based prodrugs as a means to improve the aqueous solubility of HIV inhibitors, and the introduction of the substrate envelope hypothesis as a new approach for overcoming HIV drug resistance. Finally, we discuss future directions for research, including opportunities for exploitation of novel antiretroviral targets, and the strategy of activation of latent HIV reservoirs as a means to eradicate the virus.
Co-reporter:Heng Zhang, Dongwei Kang, Boshi Huang, Na Liu, Fabao Zhao, Peng Zhan, Xinyong Liu
European Journal of Medicinal Chemistry 2016 Volume 114() pp:65-78
Publication Date(Web):23 May 2016
DOI:10.1016/j.ejmech.2016.02.051
•We reviewed recent advances on small molecular CXCR4 antagonists.•We focused on the evolution of CXCR4 and structure-activity relationships of representative CXCR4 antagonists.•We introduced the medicinal chemistry strategies and novel methodologies.CXCR4 plays vital roles in HIV-1 life cycle for it's essential in mediating the interaction of host and virus and completing the entry process in the lifecycle of HIV-1 infection. Compared with some traditional targets, CXCR4 provides a novel and less mutated drug target in the battle against AIDS. Its antagonists have no cross resistance with other antagonists. Great achievements have been made recent years and a number of small molecular CXCR4 antagonists with diversity scaffolds have been discovered. In this review, recent advances in the discovery of CXCR4 antagonists with special attentions on their evolution and structure-activity relationships of representative CXCR4 antagonists are described. Moreover, some classical medicinal chemistry strategies and novel methodologies are also introduced.Recent advances in the discovery of CXCR4 antagonists with special attentions on their evolution and structure-activity relationships of representative CXCR4 antagonists.
Co-reporter:Wenmin Chen, Peng Zhan, Dirk Daelemans, Jiapei Yang, Boshi Huang, Erik De Clercq, Christophe Pannecouque, Xinyong Liu
European Journal of Medicinal Chemistry 2016 Volume 121() pp:352-363
Publication Date(Web):4 October 2016
DOI:10.1016/j.ejmech.2016.05.054
•Piperidyl or morpholinyl group modified pyridine-type DAPY derivatives were reported.•Improved anti-HIV-1 activity and water solubility were observed.•Four compounds exhibited high anti-HIV-1 (IIIB) activity in nanomolar range.•Molecular modeling confirms the binding mode.Based on the crystallographic studies of diarylpyrimidines (DAPYs), we embarked on incorporating the hydrophilic piperidyl or morpholinyl group into the known DAPY derivatives bearing the pyridine moiety as a core structure, with the double aim to exploit additional interactions with the HIV-1 NNRTI binding pocket (NNIBP), as well as to improve the compound solubility. The antiviral evaluation result show that the most potent compounds I-8b2, I-8b3, I-8b4 and I-8c3 exhibited anti-HIV-1 (IIIB) strain activity ranging from 7.4 nM to 9.4 nM (SI = 168–1283), superior to FDA-approved drugs of nevirapine (NVP), lamivudine (3TC) and delavirdine (DLV), and comparable to etravirine (ETV), zidovudine (AZT) and efavirenz (EFV). Additionally, compounds I-8c2 and I-8c3 showed moderate activity against NNRTI resistant strains baring mutations K103N and Y181C with EC50 values of 6.2 μM and 6.8 μM, respectively. Preliminary structure-activity relationships (SARs), reverse transcriptase inhibition efficacy and molecular modeling of selected compounds are also presented. These outcomes support our design hypothesis and demonstrate that the piperidyl group modified pyridine-typed DAPY derivatives are highly potent NNRTIs with improved water solubility.Pyridine-type DAPY derivatives were modified using the piperidyl or morpholinyl group to exploit the tolerant regions of RT NNRTI binding pocket.
Co-reporter:Qing Meng, Xuwang Chen, Dongwei Kang, Boshi Huang, Wenxin Li, Peng Zhan, Dirk Daelemans, Erik De Clercq, Christophe Pannecouque, Xinyong Liu
European Journal of Medicinal Chemistry 2016 Volume 115() pp:53-62
Publication Date(Web):10 June 2016
DOI:10.1016/j.ejmech.2016.02.068
•Novel DAPY derivatives were HIV-1 inhibitors with dual structural conformations.•These compounds were designed to target the entrance channel of the NNIBP.•15b was the most promising inhibitor against wt and double mutant HIV-1 strains.•Preliminary SARs and molecular simulation of these new analogs were detailed.On the basis of structure-based bioisosteric replacement and molecular hybridization strategy, a series of novel dual structural-conformation inhibitors targeting the “entrance channel” of HIV-1 NNRTIs binding pocket (NNIBP) were designed and synthesized. All of the new compounds were evaluated for their anti-HIV activities in MT-4 cells using the MTT method. Five compounds exhibited moderate to excellent potencies inhibiting wild-type (wt) HIV-1 replication with EC50 values ranging from 31.36 μM to 0.11 μM. Among them, compound 15b was identified as the most potent inhibitor with EC50 values of 0.11 μM and 2.18 μM against wt and K103N/Y181C double mutant HIV-1 strain (RES056), respectively. In addition, preliminary structure–activity relationships (SARs) and molecular simulation studies were discussed, which may provide valuable insights for further optimization.A series of novel DAPY derivatives with dual structural conformations targeting the entrance channel of the NNRTI binding pocket were identified as inhibitors of wild-type and double mutant HIV-1 strains.
Co-reporter:Jiapei Yang, Wenmin Chen, Dongwei Kang, Xueyi Lu, Xiao Li, Zhaoqiang Liu, Boshi Huang, Dirk Daelemans, Christophe Pannecouque, Erik De Clercq, Peng Zhan, Xinyong Liu
European Journal of Medicinal Chemistry 2016 Volume 109() pp:294-304
Publication Date(Web):15 February 2016
DOI:10.1016/j.ejmech.2015.11.039
•Novel 6-substituted diarylpyridine derivatives were identified as HIV-1 inhibitors.•If, Ia and IIa exhibited anti-HIV-1 (IIIB) activity with EC50 values of 35 nM, 43 nM and 41 nM, respectively.•IIb inhibited the K103N mutation with an EC50 value of 49 nM.•Preliminary SARs and molecular simulation of these new analogues were detailed.The development of novel NNRTIs with activity against variants of HIV-1RT is crucial for overcoming treatment failure. In the present study, a series of novel 6-substituted diarylpyridine derivatives targeting the entrance channel of the NNIBP of RT were designed through a molecular hybridization strategy. Encouragingly, these new diarylpyridine derivatives were found to be active against wild-type (WT) HIV-1 with an EC50 values ranging from 0.035 μM to 1.99 μM. Nearly half of them exhibited more potent inhibitory activities in cellular assays than the control drug nevirapine (NVP). Notably, three most promising compounds If (EC50 = 35 nM), Ia (EC50 = 43 nM) and IIa (EC50 = 41 nM) showed high potency against WT and were comparable to the reference drug delavirdine (DLV) (EC50 = 33 nM). Moreover, compounds Ib, IIb and IIh displayed effective activity against the most common clinically observed single and double-mutated HIV-1 strains in micromolar concentrations. In particular, the inhibition of IIb against the K103N mutation (EC50 = 49 nM), which confers resistance to a wide variety of NNRTIs, was about 140 times more effective than NVP (EC50 = 6.78 μM), 50 times more than DLV (EC50 = 2.48 μM) and about 3 times more than EFV (EC50 = 0.12 μM), indicating that the newly designed compounds have great potential to be further developed as new anti-HIV-1 agents. Preliminary structure-activity relationships (SARs) and molecular modeling of the new diarylpyridine derivatives were discussed in detail.Through structure-based boisosteric replacement and molecular hybridization, a series of novel 6-substituted diarylpyridine derivatives were designed, synthesized and identified as inhibitors of wild-type HIV-1 strain and mutant strains in this paper.
Co-reporter:Haiyong Jia, Fuxiang Bai, Na Liu, Xiaohong Liang, Peng Zhan, Chunhong Ma, Xuemei Jiang, Xinyong Liu
European Journal of Medicinal Chemistry 2016 Volume 123() pp:202-210
Publication Date(Web):10 November 2016
DOI:10.1016/j.ejmech.2016.07.048
•26 pyrazole derivatives were prepared.•Novel pyrazole derivatives were identified as non-nucleoside HBV inhibitors.•Compound 6a3 potently suppressed the secretion of HBsAg and HBeAg.•Preliminary SARs of these new derivatives were detailed.In continuation of our efforts toward the discovery of potent non-nucleoside hepatitis B virus (HBV) inhibitors with novel structures, we have employed bioisosterism and hybrid pharmacophore-based strategy to explore the chemically diverse space of bioactive compounds. In this article, the original thiazole platform was replaced with pyrazole scaffold to yield the optimal pharmacophore moieties in order to generate novel non-nucleoside HBV inhibitors with desirable potency. Some of the new compounds were able to inhibit HBV activity in the low micromolar range. In particular, compound 6a3 displayed the most potent activity against the secretion of HBsAg and HBeAg with IC50 of 24.33 μM and 2.22 μM, respectively. The preliminary structure-activity relationship (SAR) of this new series of compounds was investigated, which may help designing more potent molecules.A series of novel pyrazole derivatives were identified as non-nucleoside HBV inhibitors via bioisosterism and pharmacophore hybrid strategy.
Co-reporter:Xueyi Lu, Xiao Li, Jiapei Yang, Boshi Huang, Dongwei Kang, Fabao Zhao, Zhongxia Zhou, Erik De Clercq, Dirk Daelemans, Christophe Pannecouque, Peng Zhan, Xinyong Liu
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 18) pp:4424-4433
Publication Date(Web):15 September 2016
DOI:10.1016/j.bmc.2016.07.041
•A series of novel purinylthioacetanilide derivatives were identified as potential HIV-1 NNRTIs.•LAD-8 displayed anti-HIV activity with an EC50 value of 0.78 μM.•LBD-6 showed moderate activity against L100I mutant and double mutant strain RES056.•Preliminary SARs of these new analogues were discussed in detail.By means of structure-based bioisosterism approach, a series of novel purinylthioacetanilide derivatives were designed, synthesized and evaluated as potent HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs). Some of the tested compounds were found to be active against wild-type (WT) HIV-1(IIIB) with EC50 in the range of 0.78–4.46 μM. Among them, LAD-8 displayed the most potent anti-HIV activity (EC50 = 0.78 μM, SI = 24). In addition, LBD-6 showed moderate activity against L100I mutant (EC50 = 5.64 μM) and double mutant strain RES056 (EC50 = 22.24 μM). Preliminary structure–activity relationships (SARs) were discussed in detail. Molecular modeling study was used to predict the optimal conformation in the NNRTI binding site, which may play a guiding role in further rational optimization.
Co-reporter:Boshi Huang;Xiao Li;Peng Zhan;Erik De Clercq;Dirk Daelemans;Christophe Pannecouque
Chemical Biology & Drug Design 2016 Volume 87( Issue 2) pp:283-289
Publication Date(Web):
DOI:10.1111/cbdd.12657
Through a structure-based molecular hybridization and bioisosterism approach, a series of novel 2-(pyridin-3-yloxy)acetamide derivatives were designed, synthesized, and evaluated for their anti-HIV activities in MT-4 cell cultures. Biological results showed that three compounds (Ia, Ih, and Ij) exhibited moderate inhibitory activities against wild-type (wt) HIV-1 strain (IIIB) with EC50 values ranging from 8.18 μm to 41.52 μm. Among them, Ij was the most active analogue possessing an EC50 value of 8.18 μm. To further confirm the binding target, four compounds were selected to implement an HIV-1 RT inhibitory assay. In addition, preliminary structure–activity relationship (SAR) analysis and some predicted physicochemical properties of three active compounds Ia, Ih, and Ij were discussed in detail. Molecular docking studies were also carried out to investigate the binding modes of Ij and the lead compound GW678248 in the binding pocket of RT, which provided beneficial information for further rational design of non-nucleoside reverse transcriptase inhibitors.
Co-reporter:Xiao Li;Boshi Huang;Zhongxia Zhou;Ping Gao;Christophe Pannecouque;Dirk Daelemans;Erik De Clercq;Peng Zhan
Chemical Biology & Drug Design 2016 Volume 88( Issue 2) pp:241-253
Publication Date(Web):
DOI:10.1111/cbdd.12751
With the continuation of our unremitting efforts toward the discovery of potent HIV-1 NNRTIs, a series of novel imidazo[4,5-b]pyridin-2-ylthioacetanilides were designed, synthesized, and evaluated for their antiviral activities through combining bioisosteric replacement and structure-based drug design. Almost all of the title compounds displayed moderate to good activities against wild-type (wt) HIV-1 strain with EC50 values ranging from 0.059 to 1.41 μm in a cell-based antiviral assay. Thereinto, compounds 12 and 13 were the most active two analogues possessing an EC50 value of 0.059 and 0.073 μm against wt HIV-1, respectively, which was much more effective than the control drug nevirapine (EC50 = 0.26 μm) and comparable to delavirdine (EC50 = 0.038 μm). In addition, one selected compound showed a remarkable reverse transcriptase inhibitory activity compared to nevirapine and etravirine. In the end of this manuscript, preliminary structure–activity relationships (SARs) and molecular modeling studies were detailedly discussed, which may provide valuable insights for further optimization.
Co-reporter:Wenxin Li;Boshi Huang;Dongwei Kang;Erik De Clercq;Dirk Daelemans;Christophe Pannecouque;Peng Zhan
Chemical Biology & Drug Design 2016 Volume 88( Issue 3) pp:380-385
Publication Date(Web):
DOI:10.1111/cbdd.12765
A series of novel 5-alkyl-6-Adamantylmethylpyrimidin-4(3H)-ones bearing various substituents at the C-2 position of the pyrimidinone ring were synthesized using a facile route and evaluated for their anti-HIV activity in MT-4 cells. The biological results demonstrated that the majority of the newly designed compounds possessed moderate efficiency in inhibiting the replication of the wild-type (WT) HIV-1 strain (IIIB) with EC50 values in the range from 0.10 to 5.39 μm. Among them, 5b1 and 5b3 proved to be the two most active inhibitors against WT HIV-1 with EC50 values of 0.10 and 0.12 μm, respectively, which were more active than nevirapine (NVP) in the same assay. In addition, HIV-1 reverse-transcriptase (RT) inhibition assay indicated that the representative compound 5b1 showed affinity to WT HIV-1 RT, and inhibited the activity of RT with an IC50 value superior to the reference drug NVP. Moreover, the preliminary structure–activity relationship (SAR) and the molecular modeling analysis of these new derivatives are also discussed.
Co-reporter:Dongwei Kang, Heng Zhang, Zhongxia Zhou, Boshi Huang, Lieve Naesens, Peng Zhan, Xinyong Liu
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 21) pp:5182-5186
Publication Date(Web):1 November 2016
DOI:10.1016/j.bmcl.2016.09.071
•First discovery of novel 3-hydroxy-quinazoline-2,4(1H,3H)-diones as specific anti-vaccinia and adenovirus agents.•24b11 displayed the most potent inhibitory activity against vaccinia with an EC50 value of 1.7 μM.•24b13 was the most potent compound against adenovirus-2 with an EC50 value of 6.2 μM.•Preliminary SARs of these novel analogues were detailed.A series of 1,2,3-triazolyl 3-hydroxy-quinazoline-2,4(1H,3H)-diones was constructed utilizing Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC) method. The biological significance of the novel synthesized quinazolines was highlighted by evaluating them in vitro for antiviral activity, wherein several compounds exhibited excellent activity specifically against vaccinia and adenovirus. Especially, 24b11 displayed the most potent inhibitory activity against vaccinia with an EC50 value of 1.7 μM, which was 15 fold than that of the reference drug Cidofovir (EC50 = 25 μM). 24b13 was the most potent compound against adenovirus-2 with an EC50 value of 6.2 μM, which proved lower than all the reference drugs. Preliminary structure–activity relationships were also discussed. To the best of our knowledge, no data are present in the literature on antiviral activity of 3-hydroxy-quinazoline-2,4(1H,3H)-diones against DNA-viruses. Thus, these findings warrant further investigations (library expansion and compound refinement) on this novel class of antiviral agents.Download high-res image (189KB)Download full-size image
Co-reporter:Peng Zhan; Yukihiro Itoh; Takayoshi Suzuki
Journal of Medicinal Chemistry 2015 Volume 58(Issue 19) pp:7611-7633
Publication Date(Web):June 18, 2015
DOI:10.1021/acs.jmedchem.5b00229
Currently, the creation of class- and isoform-selective modulators of biologically important targets is a particularly challenging problem because different isoforms within a protein family often show striking similarity in spatial quaternary structure, especially at the catalytic sites or binding pockets. Therefore, an understanding of both the precise three-dimensional structure of the target protein and the mechanisms of action of modulators is important for developing more effective and selective agents. In this Perspective, we discuss currently available rational design strategies for obtaining class- and isoform-selective inhibitors and we illustrate these strategies with the aid of specific examples from the recent literature. The strategies covered include: (1) target-derived (-dependent) de novo drug discovery methodologies, and (2) follow-on derivatization approaches from initially identified active molecules (hit-to-lead and lead-to-candidate efforts). We also comment on prospects for further development and integration of strategies to achieve target-specific or isoform-selective inhibition.
Co-reporter:Wenxin Li, Xiao Li, Erik De Clercq, Peng Zhan, Xinyong Liu
European Journal of Medicinal Chemistry 2015 Volume 102() pp:167-179
Publication Date(Web):18 September 2015
DOI:10.1016/j.ejmech.2015.07.043
•Arylthioacetanilide family proved to be highly active HIV-1 NNRTIs.•The discovery and development of the arylthioacetanilides were reviewed.•Crystal structure analyses and SARs of arylthioacetanilides were detailed.•The strategies to overcome drug resistance in arylthioacetanilides were described.The poor pharmacokinetics, side effects and particularly the rapid emergence of drug resistance compromise the efficiency of the clinically used anti-HIV drugs. Therefore, the discovery of novel and effective NNRTIs is still an extremely primary mission. Arylthioacetanilide family is one of the highly active HIV-1 NNRTIs against wide-type (WT) HIV-1 and a wide range of drug-resistant mutant strains. Especially, VRX-480773 and RDEA806 have been chosen as candidates for further clinical studies. In this article, we review the discovery and development of the arylthioacetanilides, and, especially, pay much attention to the structural modifications, SARs conclusions and molecular modeling. Moreover, several medicinal chemistry strategies to overcome drug resistance involved in the optimization process of arylthioacetanilides are highlighted, providing valuable clues for further investigations.Discovery and development of the arylthioacetanilide derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors were reviewed.
Co-reporter:Boshi Huang, Xin Liang, Cuicui Li, Wenmin Chen, Tao Liu, Xiao Li, Yueyue Sun, Lu Fu, Huiqing Liu, Erik De Clercq, Christophe Pannecouque, Peng Zhan, Xinyong Liu
European Journal of Medicinal Chemistry 2015 Volume 93() pp:330-337
Publication Date(Web):26 March 2015
DOI:10.1016/j.ejmech.2015.02.022
•Novel imidazo[1,2-a]pyrazines were identified as HIV-1 inhibitors.•4a and 5a exhibited anti-HIV-1 (IIIB) activity with EC50 values of 0.26 μM and 0.32 μM respectively.•Some drug-like properties of 4a and 5a were predicted.•Preliminary SARs and molecular simulation of these new analogues were detailed.Through a structure-guided core-refining approach, a series of novel imidazo[1,2-a]pyrazine derivatives were designed, synthesized and evaluated as HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs). Biological results of antiviral assay in MT-4 cell cultures showed that 12 target compounds displayed moderate activities against wild-type (wt) HIV-1 strain (IIIB) with EC50 values ranging from 0.26 μM to 19 μM. Among them, 4a and 5a were found to be the two most active analogues possessing EC50 values of 0.26 μM and 0.32 μM respectively, comparable to delavirdine (DLV, EC50 = 0.54 μM) and nevirapine (NVP, EC50 = 0.31 μM) in a cell-based assay. Additionally, 9 compounds showed RT inhibitory activity superior to that of NVP. Moreover, some predicted drug-like properties of representative compounds 4a and 5a, as well as the structure-activity relationship (SAR) analysis were discussed in detail. The binding mode of compound 4a was investigated by molecular simulation studies.Through a structure-guided core-refining approach, a series of novel imidazo[1,2-a]pyrazine derivatives were designed, synthesized and identified as inhibitors of wild-type HIV-1 strain in this paper.
Co-reporter:Boshi Huang, Cuicui Li, Wenmin Chen, Tao Liu, Mingyan Yu, Lu Fu, Yueyue Sun, Huiqing Liu, Erik De Clercq, Christophe Pannecouque, Jan Balzarini, Peng Zhan, Xinyong Liu
European Journal of Medicinal Chemistry 2015 Volume 92() pp:754-765
Publication Date(Web):6 March 2015
DOI:10.1016/j.ejmech.2015.01.042
•Novel [1,2,4]triazolo[1,5-a]pyrimidines were identified as potent HIV-1 inhibitors.•7d exhibited anti-HIV-1 activity with EC50 values of 8.1 nM (wt) and 13 μM (RES056).•Log P and water solubility of 7d were measured.•Preliminary SARs and molecular simulation of these new analogs were detailed.In our arduous efforts to develop new potent HIV-1 non-nucleoside reverse transcriptase (RT) inhibitors (NNRTIs), novel piperidine-linked [1,2,4]triazolo[1,5-a]pyrimidine derivatives were designed, synthesized and evaluated for their antiviral activities in MT-4 cell cultures. Biological results showed that all of the title compounds displayed moderate to excellent activities against wild-type (wt) HIV-1 strain (IIIB) with EC50 values ranging from 8.1 nM to 2284 nM in a cell-based assay. Among them, the most promising analog 7d possessed an EC50 value of 8.1 nM against wt HIV-1, which was much more potent than the reference drugs DDI, 3 TC, NVP and DLV. Additionally, 7d demonstrated weak activity against the double mutant HIV-1 strain (K103N + Y181C), and was more efficient than NVP in a RT inhibition assay. Besides, some measured and calculated physicochemical properties of 7d, like log P and water solubility, as well as the structure–activity relationships (SARs) analysis have been discussed in detail. Furthermore, the binding mode of the active compound 7d was rationalized by molecular simulation studies.A series of novel piperidine-linked [1,2,4]triazolo[1,5-a]pyrimidine derivatives were synthesized and identified as inhibitors of HIV-1 wild-type and K103N + Y181C double mutant strains in this paper.
Co-reporter:Fabao Zhao, Na Liu, Peng Zhan, Xuemei Jiang, Xinyong Liu
European Journal of Medicinal Chemistry 2015 Volume 94() pp:218-228
Publication Date(Web):13 April 2015
DOI:10.1016/j.ejmech.2015.03.012
•Thumb site I is an especially attractive target, as compounds targeting this site enable a broad genotypic coverage.•Discovery and development of indole analogues as thumb site I inhibitors were reviewed.•Crystal structure analyses of indole analogues with thumb site I were described.•The SARs and drug-like profiles of indole analogues as thumb site I inhibitors were reviewed.Hepatitis C virus (HCV) NS5B RNA-depended-RNA-polymerase (RdRp) is an essential enzyme in HCV viral replication and has no functional equivalent in mammalian cells. Several classes of nucleoside and non-nucleoside inhibitors, targeting the different allosteric sites, have demonstrated efficacy in clinical trials. Compared to other allosteric sites, thumb site I is a more compelling allosteric target with a significant number of inhibitors in clinical trials. Among them, indole analogues are the most important series of NS5B thumb site I inhibitors with considerable antiviral activity. This review focuses on the discovery and development of indole inhibitors targeting on NS5B thumb site I. Five fundamental principles, the general structure–activity relationships (SARs) model of indole scaffold, were summarized, which could pave the way for further structural optimization of indole-based anti-HCV agents.Discovery and development of indole analogues as HCV NS5B thumb site I inhibitors were reviewed.
Co-reporter:Tao Liu;Boshi Huang;Ye Tian;Xin Liang;Hong Liu;Huiqing Liu;Peng Zhan;Erik De Clercq;Christophe Pannecouque
Chemical Biology & Drug Design 2015 Volume 86( Issue 1) pp:107-113
Publication Date(Web):
DOI:10.1111/cbdd.12468
Based on the hybridization of the privileged fragments in DABO and DAPY-typed HIV-1 NNRTIs, a novel series of 4-aminopiperidinyl-linked 3,5-disubstituted-1,2,6-thiadiazine-1,1-dione derivatives were designed, synthesized, and evaluated for their in vitro anti-HIV activities in MT-4 cells. Most of the target compounds showed weak inhibitory activity against WT HIV-1. In order to confirm the mode of action of the target compounds, representative compounds Ba8 and Bb8 were selected to perform the HIV-1 RT inhibitory assay. In this assay, Ba8 and Bb8 displayed good activity with IC50 values of 3.15 and 1.52 μm, respectively. Additionally, preliminary structure–activity relationships (SARs) analysis and molecular docking studies of newly synthesized compounds are also discussed.
Co-reporter:Lingzi Zhang;Jian Guo;Xin Liu;Huiqing Liu;Erik De Clercq;Christophe Pannecouque
Chemical Biology & Drug Design 2015 Volume 86( Issue 3) pp:333-343
Publication Date(Web):
DOI:10.1111/cbdd.12497
A series of benzoyl diarylamine/ether derivatives were designed, synthesized, and evaluated for their activity against human immunodeficiency virus (HIV) in MT-4 cells. Three compounds (3b, 5a, and 6a1) exhibited moderate activities against wild-type (wt) HIV-1 with EC50 values ranging from 11 to 56 μm. Among them, compound 5a was the most potent inhibitor with a novel chemical skeleton, affording a new lead compound for further molecular optimization. An enzyme assay was also implemented to confirm the binding target of the active compounds represented by 6a1. Molecular simulation studies on compound 5a, 6a1, and 7a4 were carried out to understand their binding mode with wt HIV-1 reverse transcriptase (RT) and provided useful information for further rational design of NNRTIs.
Co-reporter:Na Liu, Fabao Zhao, Haiyong Jia, Diwakar Rai, Peng Zhan, Xuemei Jiang and Xinyong Liu
MedChemComm 2015 vol. 6(Issue 4) pp:521-535
Publication Date(Web):16 Dec 2014
DOI:10.1039/C4MD00521J
Hepatitis B is an infectious inflammatory disease of the liver, which is caused by the hepatitis B virus (HBV). Nowadays, the dramatic development of new HBV inhibitors is focused on discovering diverse non-nucleoside compounds with either novel structures or new mechanisms of action. In this review, we focus on the recent advances in discovery, structural modifications and biological activities studies of several distinct classes of synthetic non-nucleoside small molecular compounds with new mechanisms.
Co-reporter:Xin Liu;Xuwang Chen;Lingzi Zhang;Peng Zhan
Medicinal Chemistry Research 2015 Volume 24( Issue 8) pp:3314-3326
Publication Date(Web):2015 August
DOI:10.1007/s00044-015-1381-1
In this paper, 65 small molecules of HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) were aligned by two different alignment methods (substructure-based and docking-based alignment) for the construction of three-dimensional quantitative structure–activity relationship models. And the molecular descriptors were calculated by comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) methods. Statistical parameters derived from the models using the above two different alignment methods showed that the docking-based CoMFA and CoMSIA models were better than the substructure-based models with both higher internal consistencies (q2 of 0.727 and 0.698, respectively) and external predictive powers (\(r_{\text{pred}}^{2}\) of 0.906 and 0.940, respectively). According to the generated contour maps, several key structural features required for increasing the biological activity of compounds were achieved. Additionally, a docking study was also performed to explore the binding mechanisms of this series of compounds, and the obtained binding interactions are in agreement with the results of the contour maps. These structural-based and ligand-based computational studies can offer useful references for the further rational design of HIV-1 NNRTIs.
Co-reporter:Yuanchao Xie ; Dongqing Xu ; Bing Huang ; Xiuli Ma ; Wenbao Qi ; Fangyuan Shi ; Xinyong Liu ; Yingjie Zhang ;Wenfang Xu
Journal of Medicinal Chemistry 2014 Volume 57(Issue 20) pp:8445-8458
Publication Date(Web):September 25, 2014
DOI:10.1021/jm500892k
To discover group-1-specific neuraminidase (NA) inhibitors that are especially involved in combating the H5N1 virus, two series of oseltamivir derivatives were designed and synthesized by targeting the 150-cavity. Among these, compound 20l was the most potent N1-selective inhibitor, with IC50 values of 0.0019, 0.0038, and 0.0067 μM against NAs from three H5N1 viruses. These values are better than those of oseltamivir carboxylate. Compound 32 was another potent N1-selective inhibitor that exhibited a 12-fold increase in activity against the H274Y mutant relative to oseltamivir carboxylate. Molecular docking studies revealed that the 150-cavity was an auxiliary binding site that may contribute to the high selectivity of these compounds. The present work is a significant breakthrough in the discovery of potent group-1-specific neuraminidase inhibitors, which may be further investigated for the treatment of infection by the H5N1 virus.
Co-reporter:Jun Wang, Peng Zhan, Zhenyu Li, Huiqing Liu, Erik De Clercq, Christophe Pannecouque, Xinyong Liu
European Journal of Medicinal Chemistry 2014 Volume 76() pp:531-538
Publication Date(Web):9 April 2014
DOI:10.1016/j.ejmech.2014.02.047
•Synthesize a series of substituted nitropyridine derivatives as NNRTIs.•The proposed binding mode was investigated by molecular docking.•A preliminary structure–activity relationship was discussed.As a continuation of our efforts to discover and develop back-up analogs of DAPYs, novel substituted nitropyridine derivatives were designed via a structure-based core refining approach, synthesized and evaluated for their in vitro HIV-1 activity in MT-4 cells. Preliminary biological evaluation indicated that most of the compounds exhibited marked inhibitory activity against wild-type HIV-1 IIIB. Most notably, the compound 7b was identified as the most promising candidate in inhibiting HIV-1 replication with an EC50 value of 0.056 μM and a selective index (SI) of 1251, which were much better than those of NVP (EC50 = 0.23 μM) and DLV (EC50 = 0.51 μM). Some other compounds, 7k, 7c, 7j and 7e, were also endowed with a favorable anti-HIV-1 potency (EC50 = 0.034, 0.11, 0.11 and 0.16 μM, respectively). Some antivirally active compounds also showed moderate inhibitory activity against RT. Preliminary structure–activity relationships (SARs) and molecular modeling of these new analogs provide valuable avenues for future molecular optimization.A series of substituted nitropyridine derivatives were identified as potent HIV NNRTIs via a structure-based core refining approach.
Co-reporter:Zhaoqiang Liu, Wenmin Chen, Peng Zhan, Erik De Clercq, Christophe Pannecouque, Xinyong Liu
European Journal of Medicinal Chemistry 2014 Volume 87() pp:52-62
Publication Date(Web):24 November 2014
DOI:10.1016/j.ejmech.2014.09.054
•Novel DANAs derivatives were designed by a molecular hybridization approach.•The DANAs were designed to target the entrance channel of HIV-1 NNIBP.•6b11 has EC50 value of 27 nM against WT HIV-1 and SI > 12518.•6b5 has EC50 values of 29 nM and 6.1 μM against WT and mutant HIV-1 strains.•SARs were discussed in detail.Through a structure-based molecular hybridization approach, a novel series of diarylnicotinamide derivatives (DANAs) targeting the entrance channel of HIV-1 NNRTIs binding pocket (NNIBP) were rationally designed, synthesized and evaluated for their anti-HIV activities in MT-4 cells together with the inhibition against the reverse transcriptase (RT) in an enzymatic assay. Encouragingly, most of the new DANAs were found to be active against wild-type HIV-1 with an EC50 in the range of 0.027–4.54 μM. Among them, compound 6b11 (EC50 = 0.027 μM, SI > 12518) and 6b5 (EC50 = 0.029 μM, SI = 2471) were identified as the most potent inhibitors, which were more potent than the reference drugs nevirapine (EC50 = 0.31 μM) and delavirdine (EC50 = 0.66 μM). Some DANAs were also active at micromolar concentrations against the K103N + Y181C resistant mutant. Compound 6b11 exhibited the highest enzymatic inhibition activity (IC50 = 20 nM), which is equal to that of efavirenz (EC50 = 20 nM) and 31 times higher than that of nevirapine (EC50 = 0.62 μM). Preliminary structure-activity relationships (SARs) and molecular modeling of these new DANAs have been discussed.A novel series of diarylnicotinamide derivatives (DANAs) were rationally designed synthesized and evaluated as inhibitors of HIV-1 wild-type and double mutant (K103N + Y181C) RES056 strains.
Co-reporter:Liu Wang, Ye Tian, Wenmin Chen, Hong Liu, Peng Zhan, Dongyue Li, Huiqing Liu, Erik De Clercq, Christophe Pannecouque, Xinyong Liu
European Journal of Medicinal Chemistry 2014 Volume 85() pp:293-303
Publication Date(Web):6 October 2014
DOI:10.1016/j.ejmech.2014.07.104
•[1,2,4]Triazolo[1,5-a]pyrimidines were identified as effective HIV-1 inhibitors.•7n show anti-HIV-1 activity with EC50 of 0.02 μM (wt) and 7.6 μM (RES056).•Preliminary SARs and molecular modeling of these new analogues were detailed.Guided by crystal structures of HIV-1 RT/DAPY complex and molecular modeling studies, a series of novel [1,2,4]triazolo[1,5-a]pyrimidine derivatives were rationally designed via structure-based core refining approach, synthesized through the readily accessible synthetic methods and evaluated for their anti-HIV activities in MT-4 cells. Preliminary biological evaluation indicated that most of the compounds exhibited marked inhibitory activity against the wild-type HIV-1 IIIB. Particularly, compound 7n was the most potent inhibitor against wild-type and K103N/Y181C double resistant mutant strain of HIV-1, possessing EC50 values of 0.02 μM and 7.6 μM, respectively, which were much better than or similar to nevirapine (NVP, EC50 = 0.15 μM, 2.9 μM) and delavirdine (DLV, EC50 = 0.07 μM, >36 μM). Besides, some other compounds, 5b, 7c, 7e, 7f, and 7m, were also endowed with favorable anti-HIV-1 potency (EC50 = 0.07, 0.05, 0.05, 0.07, and 0.05 μM, respectively), which were better than or similar to those of NVP and DLV, suggesting a high potential to further develop this type of bridgehead nitrogen heterocycle as a novel class of NNRTIs with improved antiviral efficacy and resistance profile. The selected compound, 7i, was found moderately inhibitory towards RT (IC50 = 0.39 μM), which was higher than for ETV (IC50 = 0.56 μM). Preliminary structure–activity relationships (SARs) and molecular modeling of these new analogues were detailed in this manuscript.A series of novel [1,2,4]triazolo[1,5-a]pyrimidine derivatives were synthesized and evaluated as inhibitors of the HIV-1 wild-type and double mutant (K103N + Y181C) RES056 strains in this article.
Co-reporter:Wenmin Chen, Peng Zhan, Diwakar Rai, Erik De Clercq, Christophe Pannecouque, Jan Balzarini, Zhongxia Zhou, Huiqing Liu, Xinyong Liu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 6) pp:1863-1872
Publication Date(Web):15 March 2014
DOI:10.1016/j.bmc.2014.01.054
Based on crystallographic overlays of the known inhibitors TMC125 and R221239 complexed in RT, we designed a novel series of 4-phenoxy-6-(phenylamino)pyridin-2(1H)-one derivatives as HIV NNRTIs by molecular hybridization approach. The biological testing results indicated that 2-pyridone scaffold of these inhibitors was indispensable for their anti-HIV-1 activity, and substitution of halogen at the 3-position of the 2-pyridone ring would decrease the anti-HIV activity. Four most potent compounds had anti-HIV-1 IIIB activities at low micromolar concentrations (EC50 = 0.15–0.84 μM), comparable to that of nevirapine and delavidine. Some compounds were selected to test their anti-HIV-1 RT inhibitory action and to perform molecular modeling studies to predict the binding mode of these 2-pyridone derivatives.
Co-reporter:Ye Tian, Deping Du, Diwakar Rai, Liu Wang, Huiqing Liu, Peng Zhan, Erik De Clercq, Christophe Pannecouque, Xinyong Liu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 7) pp:2052-2059
Publication Date(Web):1 April 2014
DOI:10.1016/j.bmc.2014.02.029
In our continuous efforts to identify novel potent HIV-1 NNRTIs, a novel class of 5,7-disubstituted pyrazolo[1,5-a]pyrimidine derivatives were rationally designed, synthesized and evaluated for their anti-HIV activities in MT4 cell cultures. Biological results showed that most of the tested compounds displayed excellent activity against wild-type HIV-1 with a wide range of EC50 values from 5.98 to 0.07 μM. Among the active compounds, 5a was found to be the most promising analogue with an EC50 of 0.07 μM against wild-type HIV-1 and very high selectivity index (SI, 3999). Compound 5a was more effective than the reference drugs nevirapine (by 2-fold) and delavirdine (by 2-fold). In order to further confirm their binding target, an HIV-1 RT inhibitory assay was also performed. Furthermore, SAR analysis among the newly synthesized compounds was discussed and the binding mode of the active compound 5a was rationalized by molecular modeling studies.A novel class of 5,7-disubstituted pyrazolo[1,5-a]pyrimidine derivatives were rationally designed, synthesized and evaluated for their anti-HIV activities in MT4 cell cultures.
Co-reporter:Lingzi Zhang, Peng Zhan, Xuwang Chen, Zhenyu Li, Zhoumeng Xie, Tong Zhao, Huiqing Liu, Erik De Clercq, Christophe Pannecouque, Jan Balzarini, Xinyong Liu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 1) pp:633-642
Publication Date(Web):1 January 2014
DOI:10.1016/j.bmc.2013.10.033
A series of novel N-arylmethyl substituted piperidine-linked aniline derivatives were designed, synthesized and evaluated for their anti-HIV activity in MT-4 cells. All the new compounds showed moderate to potent activities against wild-type (wt) HIV-1 with an EC50 range from 0.022 to 2.1 μM. Among them, compound 5a6 (EC50 = 0.022 ± 0.0091 μM, SI >10,770) was confirmed to be the most potent and selective inhibitor, which proved more potent than DDI and DLV in a cell-based assay against wt HIV-1, and more efficient than NVP in an RT (reverse transcriptase) assay. Besides, it is worth noting that compound 7a1 retained moderate inhibitory activity (EC50 = 4.8 ± 0.95 μM) against the HIV-1 double RT mutant strain (K103N/Y181C). The preliminary structure–activity relationship was discussed and rationalized by molecular simulation.
Co-reporter:Diwakar Rai;Wenmin Chen;Peng Zhan;Hong Liu;Ye Tian;Xin Liang;Erik De Clercq;Christophe Pannecouque;Jan Balzarini
Chemical Biology & Drug Design 2014 Volume 84( Issue 4) pp:420-430
Publication Date(Web):
DOI:10.1111/cbdd.12328
A series of 4-(naphthalen-1-yl)-1,2,5-thiadiazol-3-hydroxyl derivatives (Ia–Im and IIa–IIe) designed as novel HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) was synthesized via an expeditious route and evaluated for their anti-HIV activities in MT-4 cell cultures. All the synthesized compounds were structurally confirmed by spectral analyses. Biological results showed that three analogues displayed moderate inhibitory activity against wild-type (wt) HIV-1 replication with EC50 values ranging from 16 to 22 μm. Molecular docking of compound Ih with wt HIV-1 RT was performed to understand the binding mode between these inhibitors and the wt HIV-1 RT and to rationalize some SARs.
Co-reporter:Peng Zhan, Yu'ning Song, Yukihiro Itoh, Takayoshi Suzuki and Xinyong Liu
Molecular BioSystems 2014 vol. 10(Issue 11) pp:2783-2799
Publication Date(Web):28 Aug 2014
DOI:10.1039/C4MB00385C
Human tankyrases 1 and 2 (TNKS1/2) are attractive pharmacological biotargets, especially for the treatment of specific types of cancer. This article provides a fairly comprehensive overview of the structural biology of the TNKS–inhibitor complex and the current medicinal chemistry strategies being used in the structure-based rational design of tankyrase-specific inhibitors.
Co-reporter:Hongfei Chen, Guoning Li, Peng Zhan, Hong Li, Shouxun Wang and Xinyong Liu
MedChemComm 2014 vol. 5(Issue 6) pp:711-718
Publication Date(Web):12 Mar 2014
DOI:10.1039/C4MD00022F
A series of novel trimethylpyrazine-2-carbonyloxy-cinnamic acids and esters were designed, synthesized and evaluated for their inhibitory effect on adenosine diphosphate (ADP)-induced platelet aggregation in vitro and also assayed for their protective effect against hydrogen peroxide (H2O2)-induced oxidative damage on Ea.hy926 cells. The results showed that many compounds exhibited high activity in one or both of the assays, of which, compound F′10 displayed the highest protective effect on the proliferation of the damaged Ea.hy926 cells (EC50 = 1.7 μM), presenting almost 40 times higher potency than that of lipoic acid, and compound F3 was the most active anti-platelet aggregation agent with IC50 = 9.6 μM, comparable to that of clopidogrel. The structure–activity relationships of these compounds were also discussed.
Co-reporter:Xiao Li, Xueyi Lu, Wenmin Chen, Huiqing Liu, Peng Zhan, Christophe Pannecouque, Jan Balzarini, Erik De Clercq, Xinyong Liu
Bioorganic & Medicinal Chemistry 2014 22(19) pp: 5290-5297
Publication Date(Web):
DOI:10.1016/j.bmc.2014.08.001
Co-reporter:Xiao Li, Wenmin Chen, Ye Tian, Huiqing Liu, Peng Zhan, Erik De Clercq, Christophe Pannecouque, Jan Balzarini, Xinyong Liu
European Journal of Medicinal Chemistry 2014 80() pp: 112-121
Publication Date(Web):
DOI:10.1016/j.ejmech.2014.04.036
Co-reporter:Wenjun Li, Peng Zhan, Erik De Clercq, Hongxiang Lou, Xinyong Liu
Progress in Polymer Science 2013 Volume 38(3–4) pp:421-444
Publication Date(Web):March–April 2013
DOI:10.1016/j.progpolymsci.2012.07.006
PEGylation, covalent attaching polyethylene glycol (PEG) polymers to therapeutic agents, is one of the most promising techniques to improve the therapeutic effect of drugs. Initially, this technology is mainly applied with macromolecular drugs, such as proteins, enzymes, with ten PEGylated biomacromolecules approved by the FDA for the treatment of related diseases. The clinical successful use of PEGylated macromolecular drug has promoted the application of this technology with small molecules drugs to overcome shortcomings associated with therapy, such as possible low solubility, high toxicity, undesirable pharmaceutical characteristics and nonspecific biodistribution profiles. So far, four PEGylated small drugs have been taken into clinical trials. This review mainly focuses on the recent advances of PEGylated small molecules, including their general configuration, and the current merits and limits of PEG modification. Herein PEG delivery systems are distinguished by therapeutic application (anti-tumor, anti-inflammatory, etc.) and their corresponding PEGylated small molecules are described in detail.
Co-reporter:Dongyue Li, Peng Zhan, Huiqing Liu, Christophe Pannecouque, Jan Balzarini, Erik De Clercq, Xinyong Liu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 7) pp:2128-2134
Publication Date(Web):1 April 2013
DOI:10.1016/j.bmc.2012.12.049
In continuation of our efforts toward identification and optimization of novel non-nucleoside reverse transcriptase inhibitors (NNRTIs), we have employed a structure-based bioisosterism strategy, with which a new series of diarylpyridazine (DAPD) derivatives were synthesized and evaluated for their anti-HIV-1 (human immunodeficiency virus type 1) activity. Most of the title compounds displayed excellent anti-HIV-1 activity at submicromolar concentrations ranging from 34 nM to 5.08 μM. The most promising compound 8g inhibited HIV-1 IIIB in MT-4 cells at a low EC50 value (0.034 μM), which was lower than the reference drug nevirapine and delavirdine. The structure activity relationships (SARs) were discussed and rationalized by docking simulations.
Co-reporter:Ye Tian;Diwakar Rai;Peng Zhan;Christophe Pannecouque;Jan Balzarini;Erik De Clercq;Huiqing Liu
Chemical Biology & Drug Design 2013 Volume 82( Issue 4) pp:384-393
Publication Date(Web):
DOI:10.1111/cbdd.12160
On the basis of structural features, binding mode, and structure–activity relationship studies of two pyrimidine-derived non-nucleoside reverse-transcriptase inhibitors, DABOs, and diaryl pyrimidines, a novel class of 1,2,6-thiadiazine-1,1-dione derivatives were rationally designed using the strategies of bioisosterism and molecular hybridization, synthesized, and evaluated for their anti-HIV activity in MT4 cell cultures. Three compounds were found to have moderate activity against HIV-1 replication with EC50 values ranging from 23 to 32 μm. To further confirm the binding target, compound IIg was selected to conduct an HIV-1 reverse-transcriptase inhibitory assay. In addition, preliminary structure–activity relationship analysis among the newly synthesized compounds was discussed, and the binding mode of the active compound IIg was rationalized by molecular docking and physicochemical studies.
Co-reporter:Wenmin Chen, Peng Zhan, Erik De Clercq, Christophe Pannecouque, Jan Balzarini, Xin Jiang, Xinyong Liu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 22) pp:7091-7100
Publication Date(Web):15 November 2013
DOI:10.1016/j.bmc.2013.09.009
A series of N2,N4-disubstituted-1,1,3-trioxo-2H,4H-pyrrolo[1,2-b][1,2,4,6]thiatriazine derivatives (PTTDs) was designed and synthesized by a facile route. The biological assay results showed that five most potent compounds displayed inhibitory activity against HIV-1 at low micromolar concentrations (EC50 = 5.1–8.9 μM). Structure–activity relationship analysis indicated that N2-(3-halogenated-benzyl) analogues were more potent than N2-(unsubstituted-benzyl) analogues. The N4-substitutions contributed to the antiviral activity in the following order: 2-/3-cyano substituted benzyl > 2-/3-halogenated benzyl > non-substituted benzyl > 4-halogenated benzyl. Docking studies of the representative compound revealed the binding conformation of these compounds and provided critical insights for the further development of PTTD analogues.
Co-reporter:Diwakar Rai, Wenmin Chen, Ye Tian, Xuwang Chen, Peng Zhan, Erik De Clercq, Christophe Pannecouque, Jan Balzarini, Xinyong Liu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 23) pp:7398-7405
Publication Date(Web):1 December 2013
DOI:10.1016/j.bmc.2013.09.051
A novel series of 3-benzyloxy-linked pyrimidinylphenylamine derivatives (8a–8s) was designed, synthesized and evaluated for their in vitro anti-HIV activity in MT-4 cell cultures. Most of the compounds inhibited wild-type (wt) HIV-1 replication in the lower micromolar concentration range (EC50 = 0.05–35 μM) with high selectivity index (SI) values (ranged from 10 to >4870). In particular, 8h and 8g displayed excellent antiretroviral activity against wt HIV-1 with low cytotoxicity (EC50 = 0.07 μM, CC50 >347 μM, SI >4870; EC50 = 0.05 μM, CC50 = 42 μM, SI = 777, respectively), comparable to that of the marked drug nevirapine (EC50 = 0.113 μM, CC50 >15 μM, SI >133). In order to confirm the binding target, 8h was selected to perform the anti-HIV-1 RT assay. Additionally, preliminary structure activity relationship (SAR) analysis and molecular docking studies of newly synthesized compounds were also discussed, as well as the predicted physicochemical properties.
Co-reporter:Hongfei Chen, Guoning Li, Peng Zhan, Xiuli Guo, Qian Ding, Shouxun Wang and Xinyong Liu
MedChemComm 2013 vol. 4(Issue 5) pp:827-832
Publication Date(Web):11 Mar 2013
DOI:10.1039/C3MD20352B
A series of novel ligustrazinylated derivatives was designed, synthesized and evaluated for their protective effects against hydrogen peroxide (H2O2)-induced oxidative damage on ECV-304 cells, and also assayed for their inhibition effects on platelet aggregation induced by adenosine diphosphate (ADP). Biological results showed that some compounds exhibited moderate to potent activities in one or both of the assays. Among these compounds, compound 3 displayed the highest protective effect on the damaged ECV-304 cells with EC50 = 0.0040 mM, and compound 1c was the most active platelet aggregation inhibitor (EC50 = 0.40 mM). Structure–activity relationships were briefly discussed.
Co-reporter:Yu’ning Song, Peng Zhan, Dongwei Kang, Xiao Li, Ye Tian, Zhenyu Li, Xuwang Chen, Wenmin Chen, Christophe Pannecouque, Erik De Clercq and Xinyong Liu
MedChemComm 2013 vol. 4(Issue 5) pp:810-816
Publication Date(Web):08 Mar 2013
DOI:10.1039/C3MD00028A
In continuation of our endeavors to develop new, potent, selective, and less toxic anti-HIV agents, we describe our structure-based bioisosterism design, synthetic strategy, and structure–activity relationship (SAR) studies that led to the identification of pyridazinylthioacetamides, a novel class of NNRTIs, isosteres of arylazolylthioacetanilide derivatives. Nearly all of the tested compounds inhibited HIV-1 strain IIIB replication in the lower micromolar concentration range (EC50: 0.046–5.46 μM). Notably, the most promising compound 8k exhibited extremely potent inhibitory activity against HIV-1 replication with an EC50 value of 0.046 μM, CC50 of 99.9 μM and the viral selectivity index amounted to 2149. These values were much better than those of NVP (EC50 = 0.09 μM) and DDC (EC50 = 1.04 μM). Compound 8k also exhibited moderate inhibition of enzymatic activity with an IC50 value of 4.06 μM, which was of the same order of magnitude as that of NVP (2.74 μM). Docking calculations were also performed to investigate the binding mode of compound 8k into the non-nucleoside binding site of HIV-1 RT and to rationalize some SARs.
Co-reporter:Wenmin Chen;Peng Zhan;Diwakar Rai;Boshi Huang;Jiapei Yang
Heteroatom Chemistry 2013 Volume 24( Issue 6) pp:495-501
Publication Date(Web):
DOI:10.1002/hc.21123
ABSTRACT
We reported a facile and efficient methodology to synthesize derivatives of pyrrolo[1,2-b][1,2,4,6]thiatriazine (PTTD): a new bridgehead nitrogen heterocyclic system. Treatment of methyl pyrrole-2-carboxylate with N-benzylsulfamoyl chlorides followed by subsequent hydrolysis reaction afforded 1-benzylsulfamoyl-1H-pyrrole-2-carboxylic acids, which underwent ring-closing reaction by treatment with diphenyl phosphorazidate via Curtius rearrangement; subsequent alkylation reaction afforded the desired 2,4-disubstituted-1,1,3-trioxo-2H,4H-PTTDs.
Co-reporter:Xuwang Chen, Xin Liu, Qing Meng, Ding Wang, Huiqing Liu, Erik De Clercq, Christophe Pannecouque, Jan Balzarini, Xinyong Liu
Bioorganic & Medicinal Chemistry Letters 2013 23(24) pp: 6593-6597
Publication Date(Web):
DOI:10.1016/j.bmcl.2013.10.059
Co-reporter:Dr. Xuwang Chen;Yuanyuan Li;Shufang Ding; Jan Balzarini; Christophe Pannecouque; Erik DeClercq;Dr. Huiqing Liu; Xinyong Liu
ChemMedChem 2013 Volume 8( Issue 7) pp:1117-1126
Publication Date(Web):
DOI:10.1002/cmdc.201300130
Abstract
In our continued efforts to discover more active and less toxic HIV-1 non-nucleoside reverse transcriptase inhibitors, we recently designed a novel series of piperidine-linked pyridine analogues on the basis of diarylpyrimidine derivatives, among which two drugs—etravirine and rilpivirine—are approved for use by the US FDA. The title compounds were evaluated for activity against wild-type and resistant mutant strains of HIV-1 as well as HIV-2 in MT-4 cells. The highly potent compound BD-c1 (EC50=10 nM, CC50≥146 μM, SI≥14 126) displays lower cytotoxicity and higher selectivity than etravirine (EC50=2.2 nM, CC50=28 μM, SI=12 884) against wild-type HIV-1. Compound BD-e2 (EC50=5.1 nM) shows greater antiviral efficacy against wild-type HIV-1 than do the four reference drugs nevirapine, delavirdine, efavirenz, and zidovudine. Many compounds were also found to be active against the frequently observed drug-resistant double mutant (K103N+Y181C) HIV-1 strain. Herein we report the design, synthesis, anti-HIV evaluation, preliminary structure–activity relationships, and molecular simulations of novel piperidine-linked pyridine analogues.
Co-reporter:Dongyue Li ; Peng Zhan ; Erik De Clercq
Journal of Medicinal Chemistry 2012 Volume 55(Issue 8) pp:3595-3613
Publication Date(Web):January 23, 2012
DOI:10.1021/jm200990c
Co-reporter:Xuwang Chen, Peng Zhan, Christophe Pannecouque, Jan Balzarini, Erik De Clercq, Xinyong Liu
European Journal of Medicinal Chemistry 2012 Volume 51() pp:60-66
Publication Date(Web):May 2012
DOI:10.1016/j.ejmech.2012.02.019
A novel series of piperidine-substituted triazine derivatives have been synthesized and evaluated for anti-HIV activities in MT-4 cells. Most compounds displayed extremely promising activity against wild-type HIV-1 with EC50 values in low nanomolar concentration, better than that of Nevirapine, Delavirdine, Zidovudine and Dideoxycitidine, and higher potency towards the resistant mutant strain K103N/Y181C than that of Nevirapine and Delavirdine. Selected compounds were also assayed against reverse transcriptase with lower IC50 values than that of Nevirapine. The structure-activity relationship (SAR) of these novel structural congeners was also discussed.The novel piperidine-substituted triazine derivatives showed extremely promising activity against wild-type HIV-1 and moderate potency towards the K103N/Y181C resistant mutant strain.Highlights► Novel piperidine-substituted triazine derivatives were synthesized. ► Promising activity against wild-type HIV-1 (EC50 = 7.0 nM, SI = 3240). ► IC50 values against HIV-1 RT were in low micromolar level.
Co-reporter:Xiao Li, Peng Zhan, Hong Liu, Dongyue Li, Liu Wang, Xuwang Chen, Huiqing Liu, Christophe Pannecouque, Jan Balzarini, Erik De Clercq, Xinyong Liu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 18) pp:5527-5536
Publication Date(Web):15 September 2012
DOI:10.1016/j.bmc.2012.07.026
In continuation of our efforts toward the discovery of potent HIV-1 NNRTIs with novel structures, we have employed a scaffold hopping strategy to explore the chemically diversed space of bioactive compounds. The original arylazolylthioacetanilide platform was replaced with different imidazopyridinyl- thioacetanilide scaffolds to yield the optimal pharmacophore moieties in order to generate novel NNRTIs with desirable potency. Some of the new compounds proved able to inhibit HIV-1 replication in the low micromolar range. In particular, compound 5b16 displayed the most potent anti-HIV-1 activity (EC50 = 0.21 ± 0.06 μM), inhibiting HIV-1 IIIB replication in MT-4 cells more effectively than dideoxycytidine (EC50 = 1.4 ± 0.1 μM) and similarly with nevirapine (EC50 = 0.20 ± 0.10 μM). Preliminary structure–activity relationship (SAR) of the newly synthesized congeners is discussed, and molecular modeling study is performed to rationalize the SAR conclusions.Graphical abstractBy means of scaffold hopping strategy, imidazopyridine was used as a new bioisostere to replace the five-membered heterocyclic lead structures. This strategy led to the identification of imidazopyridinylthioacetanilide NNRTIs with potency against HIV-1 replication in the low micromolar concentration range.
Co-reporter:Zhenyu Li;Peng Zhan;Lieve Naesens;Evelien Verlinden;Ailin Liu;Guanhua Du;Erik De Clercq
Chemical Biology & Drug Design 2012 Volume 79( Issue 6) pp:1018-1024
Publication Date(Web):
DOI:10.1111/j.1747-0285.2012.01344.x
The diversity-oriented chemistry synthesis together with the random screening approach has permitted the discovery and optimization of novel antiviral lead compounds. In this paper, a series of novel 5-substituted-2-(4-substituted phenyl)-1,3-benzoxazoles was synthesized and evaluated for their in vitro anti-influenza A virus and anti-influenza B virus activity. The activity was monitored by the MTS assay in the Madin–Darby canine kidney cells. Compound 7h showed excellent inhibitory activity and selective index against A/H3N2 (EC50 = 37.03 μm, SI > 5), which were all higher than that of the reference drug oseltamivir (EC50 > 59.00 μm, SI > 1). However, no compound displays inhibitory activity against influenza B virus.
Co-reporter:Lijuan Deng;Xiuli Guo;Li Zhai;Yuning Song;Hongfei Chen;Peng Zhan;Jingde Wu
Chemical Biology & Drug Design 2012 Volume 79( Issue 5) pp:731-739
Publication Date(Web):
DOI:10.1111/j.1747-0285.2012.01332.x
A series of novel stilbene derivatives containing ligustrazinyl moiety was designed, synthesized, and assayed for their protective effects on damaged endothelial cells. The results showed that most ligustrazinyl stilbene derivatives exhibited high protective effects on the human umbilical vascular endothelial cells (HUVECs) damaged by hydrogen peroxide in comparison with Ligustrazine. The stilbene derivatives A6, A9, A11, A21, A24, A25, and A27 exhibited high potency with low EC50 values ranged from 0.0249 μm to 0.0898 mm. Compound A27 displayed EC50 0.0249 μm, which is 30 000 times higher than that of Ligustrazine, presenting a most promising lead for further investigation. Structure–activity relationships were briefly discussed.
Co-reporter:Peng Zhan, Wenmin Chen, Zhenyu Li, Xiao Li, Xuwang Chen, Ye Tian, Christophe Pannecouque, Erik De Clercq, Xinyong Liu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 23) pp:6795-6802
Publication Date(Web):1 December 2012
DOI:10.1016/j.bmc.2012.09.058
The present work is an extension of our ongoing efforts towards the development and identification of new molecules with anti-HIV activity which have previously led to the discovery of arylazolylthioacetanilides as highly active NNRTIs. In this article, a series of 2-2-(3-(2-chlorophenyl)pyrazin-2-ylthio)-N-arylacetamide derivatives were synthesized and evaluated for in vitro anti-HIV activity. Most of the tested compounds exhibited moderate activities against wild-type HIV-1. Among them, compound 6k showed significant activity against wild-type HIV-1 with an EC50 value of 1.7 μM, along with moderate activity against wild-type reverse transcriptase (RT). The preliminary structure–activity relationship (SAR) and docking calculations of this new series of compounds were also investigated, which may help designing more potent molecules.
Co-reporter:Xuwang Chen, Peng Zhan, Xin Liu, Ziheng Cheng, Caicai Meng, Siyuan Shao, Christophe Pannecouque, Erik De Clercq, Xinyong Liu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 12) pp:3856-3864
Publication Date(Web):15 June 2012
DOI:10.1016/j.bmc.2012.04.030
A novel series of piperidine-linked amino-triazine derivatives were designed, synthesized and evaluated for in vitro anti-HIV activity as non-nucleoside reverse transcriptase inhibitors on the basis of our previous work. Screening results indicated that most compounds showed excellent activity against wild-type HIV-1 with EC50 values in low nanomolar concentration range (especially compound 6b3, EC50 = 4.61 nM, SI = 5945) and high activity against K103N/Y181C resistant mutant strain of HIV-1 with EC50 values in low micromolar concentration range. In addition, preliminary structure–activity relationship and molecular modeling of these new analogs were detailed in this manuscript.Piperidine-substituted amino-triazine derivatives were designed, introducing amino to the triazine ring and polar hydrophilic groups to the right wing, with excellent activity against wild-type HIV-1.
Co-reporter:Peng Zhan, Xiao Li, Zhenyu Li, Xuwang Chen, Ye Tian, Wenmin Chen, Xinyong Liu, Christophe Pannecouque, Erik De Clercq
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 23) pp:7155-7162
Publication Date(Web):1 December 2012
DOI:10.1016/j.bmcl.2012.09.062
The development of new HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) offers the possibility of generating novel chemical entities of increased potency. Previous investigations in our laboratory resulted in the discovery of several novel series of arylazolylthioacetanilides as potent NNRTIs. In this study, based on the structure-based bioisosterism strategy, novel 1,2,4-triazin-6-yl thioacetamide derivatives were designed, synthesized and evaluated for their anti-HIV activity in MT-4 cells. Among them, the most promising compound was 8b15 with double-digit nanomolar activity against wild-type HIV-1 (EC50 = 0.018 ± 0.007 μM) and moderate activity against the double mutant strain RES056 (EC50 = 3.3 ± 0.1 μM), which indicated that 1,2,4-triazin-6-yl thioacetamide can be used as a novel scaffold to develop a new class of potent NNRTIs active against both wild-type and drug-resistant HIV-1 strains. In addition, preliminary structure–activity relationship (SAR) and molecular modeling results are also briefly discussed, which provide some useful information for the further design of novel NNRTIs.The 1,2,4-triazin-6-ylthioacetamides were identified as potent NNRTIs designed by structure-based bioisosterism strategy. The HIV-1(IIIB) inhibitory activity of compound 8b15 (EC50 = 0.018 ± 0.007 μM) was about seventy-eight, eleven and two fold more active than that of positive control drugs DDC, NVP and DLV respectively, and displayed similar anti-HIV-1 activity as that of EFV. Because of their excellent potency, these 1,2,4-triazin-6-yl thioacetamide derivatives may have antiviral potential and should be further pursued as next-generation NNRTIs.
Co-reporter:Hongfei Chen, Guoning Li, Peng Zhan, Xinyong Liu
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 11) pp:5609-5615
Publication Date(Web):November 2011
DOI:10.1016/j.ejmech.2011.09.030
A series of novel ligustrazinyloxy-cinnamic acid derivatives were designed, synthesized and evaluated for their inhibitory effect on adenosine diphosphate (ADP)-induced platelet aggregation in vitro, and also assayed for their protective effect against hydrogen peroxide (H2O2)-induced oxidative damage on ECV-304 cells. Some compounds exhibited high activity in one or both of the assays, of which, compound 2e displayed the highest protective effect on the proliferation of the damaged ECV-304 cells (EC50 = 0.020 mM), and compound 2f was the most active anti-platelet aggregation agent (EC50 = 0.054 mM). Structure–activity relationships were briefly discussed.A series of ligustrazine derivatives were synthesized and evaluated for their inhibitory effect on platelet aggregation and protective effect against oxidative damage. The structure–activity relationships were briefly discussed.Highlights► Ligustrazine and cinnamic acids as constructing fragments. ► Inhibitory effect on platelet aggregation and protective effect against oxidative damage. ► Carboxyl group is indispensible for anti-platelet aggregation activity.
Co-reporter:Peng Zhan, Xuwang Chen, Xiao Li, Dongyue Li, Ye Tian, Wenwen Chen, Christophe Pannecouque, Erik De Clercq, Xinyong Liu
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 10) pp:5039-5045
Publication Date(Web):October 2011
DOI:10.1016/j.ejmech.2011.08.011
The development of novel HIV-1 NNRTIs offers the possibility of generating novel structures with increased potency. Based on the bioisosteric principle, a novel series of 2-(2-(2,4-dichlorophenyl)-2H-1,2,4-triazol-3-ylthio)-N-arylacetamide derivatives were designed, synthesized using a simple and efficient synthetic route, structurally confirmed by spectral analysis, evaluated for their anti-HIV activity in MT-4 cells and their inhibitory effect on HIV-1 RT. The results showed that some of the new compounds displayed low micromolar potency for inhibiting HIV-1 replication and promising activities against several selected resistant strains that confer resistance to current NNRTIs. However, all newly synthesized derivatives were not active against HIV-2 replication.Some newly synthesized 1,2,4-triazolacetamide derivatives displayed low micromolar potency for inhibiting HIV-1 replication and promising activities against several selected resistant strains that confer resistance to current NNRTIs.Highlights► A facile synthetic approach towards novel 1,2,4-triazolacetamides was described. ► More than half of the tested compounds were found to be active against HIV-1. ► Some derivatives showed promising activities against several selected resistant strains.
Co-reporter:Jing Zhang, Peng Zhan, Jingde Wu, Zhenyu Li, Yan Jiang, Weiying Ge, Christophe Pannecouque, Erik De Clercq, Xinyong Liu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 14) pp:4366-4376
Publication Date(Web):15 July 2011
DOI:10.1016/j.bmc.2011.05.024
A series of novel S-DABO analogues of 5-alkyl-2-arylthio-6-((3,4-dihydroquinolin-1(2H)-yl)methyl)pyrimidin-4(3H)-ones were synthesized and evaluated as inhibitors of human immunodeficiency virus type-1 (HIV-1). Among them, the most potent HIV-1 inhibitors were compounds 6c1,6c6, and 6b1 (EC50 = 0.24 ± 0.05, 0.38 ± 0.13, 0.39 ± 0.05 μM, respectively), which possess improved or similar HIV-1 inhibitory activity compared with nevirapine (NVP) (EC50 = 0.21 μM) and delavirdine (DLV) (EC50 = 0.32 μM). None of these compounds were active against HIV-2 replication. Furthermore, enzyme inhibitory assays were performed with selected derivatives against HIV-1 wtRT, confirming that the main target of these compounds is the HIV-1 RT and these new S-DABOs are acting as NNRTIs. The preliminary structure–activity relationship (SAR) of these new congeners is discussed briefly and rationalized by docking studies.A series of novel S-DABO analogues of 5-alkyl-2-arylthio-6-((3,4-dihydroquinolin-1(2H)-yl)methyl)pyrimidin-4(3H)-ones were synthesized and evaluated as inhibitors of human immunodeficiency virus type-1 (HIV-1).
Co-reporter:Mingyan Yu;Ailin Liu;Guanhua Du;Lieve Naesens;Evelien Verlinden;Erik De Clercq
Chemical Biology & Drug Design 2011 Volume 78( Issue 4) pp:596-602
Publication Date(Web):
DOI:10.1111/j.1747-0285.2011.01180.x
A series of novel dihydro-alkyloxy-benzyl-oxopyrimidine derivatives were synthesized and evaluated for their activity against influenza virus in Madin–Darby canine kidney cells. Four dihydro-alkyloxy-benzyl-oxopyrimidine derivatives (4a1, 4a2, 4a3, and 4d1) showed potent activity against influenza virus. Among them, compound 4a3 was the most promising lead with broad activity against influenza A (antiviral EC50 values of 9 and 18 μm for the A/H1N1 and A/H3N2 subtype, respectively) and influenza B viruses (EC50: 33 μm). The antiviral mechanism of action of these dihydro-alkyloxy-benzyl-oxopyrimidine derivatives must be quite different from that of the currently approved anti-influenza virus drugs that target the viral M2 or neuraminidase proteins. The dihydro-alkyloxy-benzyl-oxopyrimidine derivatives represent a new avenue for further optimization and development of novel anti-influenza virus agents.
Co-reporter:Dr. Mingyan Yu;Zhenyu Li;Shuai Liu;Erkang Fan;Dr. Christophe Pannecouque;Dr. Erik DeClercq;Dr. Xinyong Liu
ChemMedChem 2011 Volume 6( Issue 5) pp:826-833
Publication Date(Web):
DOI:10.1002/cmdc.201000555
Abstract
A series of new 5-alkyl-2-phenylaminocarbonylmethylthiopyrimidin-4(3H)-ones bearing variously substituted arylmethyl moieties at the C6 position of the pyrimidine ring were synthesized and evaluated for anti-HIV activity in MT-4 cells. Most of these new congeners exhibited moderate to good activities against the wild-type virus, with EC50 values in the range of 1.40–0.19 μM. Among them, 2-[(4-cyanophenylamino)carbonylmethylthio]-6-(2-chloro-6-fluorobenzyl)-5-ethylpyrimidin-4(3H)-one 4 b6 is one of the compounds endowed with the highest broad-spectrum HIV-1 inhibitory activity, with EC50 values of 0.19±0.005 μM against the wild-type virus, 1.05±0.24 μM (twofold resistance) against the E138K strain, and 2.38±0.13 μM (4.5-fold resistance) against the Y181C strain. Furthermore, reverse transcriptase (RT) inhibition assays against wild-type HIV-1 RT were performed with selected derivatives, confirming that the main target of these compounds is HIV-1 RT and that these new S-DABO analogues act as non-nucleoside RT inhibitors (NNRTIs). Structure–activity relationship and molecular modeling analyses of these new congeners are also discussed.
Co-reporter:Peng Zhan;Xin-Yong Liu;Zhen-Yu Li;Zeng-Jun Fang;Christophe Pannecouque;Erik DeClercq
Chemistry & Biodiversity 2010 Volume 7( Issue 7) pp:1717-1727
Publication Date(Web):
DOI:10.1002/cbdv.200900197
Abstract
As part of our studies to discover new HIV-1 reverse transcriptase inhibitors, a series of 3,4-dichlorophenyl substituted 1,2,3-thiadiazole thioacetanilide (TTA=[(1,2,3-thiadiazole-5-yl)sulfanyl]acetanilide) derivatives were synthesized, and in vitro anti-HIV activity was evaluated. The results revealed that nearly half of the compounds show moderate-to-good inhibitory potency against HIV-1. In particular, compound 7f is highly potent, with an EC50 value of 0.95±0.33 μM. The preliminary structure–activity relationship among the newly synthesized congeners is discussed.
Co-reporter:Dr. Wenjun Li;Yu Chang;Peng Zhan;Dr. Na Zhang;Dr. Xinyong Liu;Dr. Christophe Pannecouque;Dr. Erik DeClercq
ChemMedChem 2010 Volume 5( Issue 11) pp:1893-1898
Publication Date(Web):
DOI:10.1002/cmdc.201000352
Abstract
A poly(ethylene glycol) (PEG) conjugate of 3′-azido-3′- deoxythymidine (AZT, zidovudine) was designed and synthesized as a novel sustained-release prodrug. In the synthetic process, a succinate diester spacer was used to covalently couple AZT with methoxy poly(ethylene glycol) (mPEG; MW=2000). The conjugate was characterized by Fourier transform infrared (FTIR) and NMR spectroscopies and matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry (MS). The in vitro release was determined in hydrochloride (HCl) solution (pH 1.2) and phosphate-buffered solution (PBS; pH 6.8), which showed the release rate of AZT from the conjugate was slower than that from the free drug, suggesting its possible increased retention in gastrointestinal conditions. Pharmacokinetic properties were evaluated experimentally by oral administration in mice. Compared to free AZT, the absorption half-life (tka) and elimination half life (tβ) of AZT released from the conjugate were both extended to 0.51±0.03 h (p <0.01) and 2.94±0.24 h (p <0.01), respectively. Evaluation of the in vitro anti-HIV activities showed mPEG-AZT exhibited good inhibition of HIV-1, with an EC50 value of 0.0634 μM, but it is lower than that of free AZT. These results show that the conjugate is capable of releasing the parent drug in a sustained profile, potentially providing a feasible alternative to oral administration of AZT in a clinical setting.
Co-reporter:Peng Zhan;Hongbing Liu;Yan Wang
Medicinal Chemistry Research 2010 Volume 19( Issue 7) pp:652-663
Publication Date(Web):2010 September
DOI:10.1007/s00044-009-9220-x
A series of novel N′-arylidene-2-[1-(naphthalen-1-yl)-1H-tetrazol-5-ylthio] acetohydrazides was synthesized and evaluated, as nonnucleoside reverse transcriptase inhibitors (NNRTIs), for their in vitro HIV-1 and HIV-2 activity using the IIIB strain and ROD strain, respectively. The activity was monitored by the inhibition of the virus-induced cytopathic effect in the human T-lymphocyte (MT-4) cells. All of the new compounds were structurally confirmed by spectral analyses. Compounds 5q and 5r showed EC50 of 29.62 μM (CC50 of 169.24 ± 23.83μM) and 31.62 μM (CC50 > 309.06 μM), and resulting in selectivity index of 6 and >9, respectively. However, all newly synthesized derivatives were not active against HIV-2 replication.
Co-reporter:Peng Zhan, Xinyong Liu, Zengjun Fang, Zhenyu Li, Christophe Pannecouque, Erik De Clercq
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 11) pp:4648-4653
Publication Date(Web):November 2009
DOI:10.1016/j.ejmech.2009.06.037
A series of 2-(4-(naphthalen-2-yl)-1,2,3-thiadiazol-5-ylthio)acetamide (TTA) derivatives were synthesized and evaluated as potent inhibitors of HIV-1. Among the newly disclosed TTAs, compounds 7f, 7g and 7c were the most potent inhibitors of HIV-1 replication of the series (EC50 = 0.17 ± 0.02, 0.36 ± 0.19 and 0.39 ± 0.05 μM, respectively), coupled with a reasonable selectivity index (SI > 1452, >845, and >774, respectively). They possess improved or similar HIV-1 inhibitory activity compared with NVP (EC50 = 0.208 μM) and DLV (EC50 = 0.320 μM). The preliminary structure–activity relationships among the newly synthesized congeners are discussed briefly and rationalized by docking studies.Model of 7f docked into the RT non-nucleoside binding pocket (PDB code: 3DLG).
Co-reporter:Peng Zhan, Xinyong Liu, Junjie Zhu, Zengjun Fang, Zhenyu Li, Christophe Pannecouque, Erik De Clercq
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 16) pp:5775-5781
Publication Date(Web):15 August 2009
DOI:10.1016/j.bmc.2009.07.028
A series of 2-(1-aryl-1H-imidazol-2-ylthio)acetamide [imidazole thioacetanilide (ITA)] derivatives were synthesized and evaluated as potent inhibitors of human immunodeficiency virus type-1 (HIV-1). Among them, the most potent HIV-1 inhibitors were 4a5 (EC50 = 0.18 μM), and 4a2 (EC50 = 0.20 μM), which were more effective than the lead compound L1 (EC50 = 2.053 μM) and the reference drugs nevirapine and delavirdine. The preliminary structure–activity relationship (SAR) of the newly synthesized congeners is discussed.
Co-reporter:Peng Zhan, Xinyong Liu, Zengjun Fang, Christophe Pannecouque, Erik De Clercq
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 17) pp:6374-6379
Publication Date(Web):1 September 2009
DOI:10.1016/j.bmc.2009.07.027
The development of new HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) offers the possibility of generating novel structures of increased potency. Based on the bioisosteric principle, novel series of 1,2,3-selenadiazole thioacetanilide derivatives were designed, and synthesized using an original synthetic route, structurally confirmed by spectral analysis, and evaluated for their anti-HIV activity in MT-4 cells. The results showed that only compound 7f possessed potent activity against HIV-1 replication (EC50 = 2.45 μM) similar to the prototype series of sulfanyltriazoles. None of the compounds were active against HIV-2 replication.A series of novel 2-(4-(3,4-dichlorophenyl)-1,2,3-selenadiazol-5-ylthio)acetamides derivatives were synthesized using an original synthetic route, and tested against HIV in MT-4 cells. Compounds 7f (EC50 = 2.45 μM), 7h (EC50 = 5.42 μM), 7i (EC50 = 5.56 μM), 7k (EC50 = 6.65 μM), and 7l (EC50 = 5.59 μM), were found to be the most potent anti-HIV-1 agents of this series.
Co-reporter:Mingyan Yu, Xinyong Liu, Zhenyu Li, Shuai Liu, Christophe Pannecouque, Erik De Clercq
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 22) pp:7749-7754
Publication Date(Web):15 November 2009
DOI:10.1016/j.bmc.2009.09.035
A series of novel 2-(phenylaminocarbonylmethylthio)-6-(2,6-dichlorobenzyl)-pyrimidin-4(3H)-ones have been designed and synthesized. All of the new compounds were evaluated for their anti-HIV activities in MT-4 cells. Most of these new compounds showed moderate to potent activities against wild-type HIV-1 with an EC50 ranging from 4.48 μM to 0.18 μM. Among them, 2-[(4-bromophenylamino)carbonylmethylthio]-6-(2,6-dichlorobenzyl)-5-methylpyrimidin-4(3H)-one 4b3 was identified as the most promising compound (EC50 = 0.18 ± 0.06 μM, CC50 >243.56 μM, SI >1326). The structure–activity relationship (SAR) of these new congeners is discussed.The compound 4b3 was docked into the NNRTIs binding pocket (NNIBP) of HIV-1 RT (PDB code: 1RT2) using Autodock Vina [http://vina.scripps.edu]. The docking result of 4b3 is showed by PyMOL [http://pymol.sourceforge.net].
Co-reporter:Xian-Chao Cheng, Xin-Yong Liu, Wen-Fang Xu, Xiu-Li Guo, Ning Zhang, Yu-Ning Song
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 8) pp:3018-3024
Publication Date(Web):15 April 2009
DOI:10.1016/j.bmc.2009.03.012
A series of novel acylpiperazinyl Ligustrazine derivatives was designed, synthesized, and their protective effects on damaged ECV-304 cells and antiplatelet aggregation activities were evaluated. The results showed that compound E33 displayed most potential protective effects on the ECV-304 cells damaged by hydrogen peroxide, and compound E1 was found to be the most active antiplatelet aggregation agent. Structure–activity relationships were briefly discussed.A series of novel acylpiperazinyl Ligustrazine derivatives was designed and synthesized. Their protective effects on damaged ECV-304 cells and antiplatelet aggregation activities were reported.
Co-reporter:Peng Zhan, Xinyong Liu, Zhenyu Li, Zengjun Fang, Zhong Li, Defeng Wang, Christophe Pannecouque, Erik De Clercq
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 16) pp:5920-5927
Publication Date(Web):15 August 2009
DOI:10.1016/j.bmc.2009.07.004
A novel synthetic route and anti-HIV activity evaluation of a new series of 2-(4-(2,4-dibromophenyl)-1,2,3-thiadiazol-5-ylthio)acetamide (TTA) derivatives are described. Bioactivity assay indicated that most of the title compounds showed good activities against HIV-1. In particular, compound 7c displayed the most potent anti-HIV-1 activity (EC50 = 36.4 nM), inhibiting HIV-1 replication in MT-4 cells more effectively than NVP (by sevenfold) and DLV (by eightfold). The preliminary structure–activity relationships (SAR) of the newly synthesized congeners are discussed, and molecular modeling of compound 7c in complex with HIV-1 RT is described, allowing rationalization of some SAR conclusions.The compound 7c was built and docked into the NNRTIs binding pocket (NNIBP) of HIV-1 RT (PDB code: 3DLG) using Autodock Vina [http://vina.scripps.edu]. The docking result of 7c is showed by PyMOL.
Co-reporter:Yongqiang Lin, Xinyong Liu, Renzhang Yan, Jin Li, Christophe Pannecouque, Myriam Witvrouw, Erik De Clercq
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 1) pp:157-163
Publication Date(Web):1 January 2008
DOI:10.1016/j.bmc.2007.10.013
A series of novel 1,3-disubstituted thieno[3,2-c] [1,2,6]thiadiazin-4(3H)-one 2,2-dioxides (TTDDs), designed as non-nucleoside reverse transcriptase inhibitors (NNRTIs), was synthesized, structurally confirmed by spectral analysis and evaluated for their anti-HIV-1 activities by inhibition of HIV-1(IIIB)-induced cytopathogenicity in MT-4 cell culture. The results showed that TTDD analogues exhibited marked potency as anti-HIV-1 agents. The most active and selective compound was 1-(3-cyano)benzyl-3-benzyl-thieno[3,2-c][1,2,6]thiadiazin-4(3H)-one 2,2-dioxide (5f) with a 50% effective concentration (EC50) of 4.0 μM and a selectivity index (SI) of >76. The structure–activity relationship (SAR) is discussed.Novel TTDDs were designed, synthesized and evaluated for their anti-HIV-1 activities in cell culture, and proved to be both potent and selective as HIV-1 inhibitors. The structure–activity relationship (SAR) is discussed.
Co-reporter:Xin-yong Liu;Ren-zhang Yan;Yan Wang;Peng Zhan;Erik De Clercq;Christophe Pannecouque;Myriam Witvrouw;Maria Teresa Molina;Salvador Vega
Archiv der Pharmazie 2008 Volume 341( Issue 4) pp:216-222
Publication Date(Web):
DOI:10.1002/ardp.200700216
Abstract
A series of novel 2,4-disubstituted 7-methyl-1,1,3-trioxo-2,4-dihydro-pyrazolo[4,5-e] [1,2,4]thiadiazines (PTDs) was synthesized, structurally confirmed by spectral analysis, and evaluated for their anti-HIV activities by inhibition of HIV-induced cytopathogenicity in MT-4 cell culture. The results showed that some compounds exhibited inhibitory activity specifically against HIV-2 replication. The most active HIV-2 inhibitor was compound 7i (R1 = benzyl, R2 = 4-t-butyl-benzyl) with an EC50 value of 18.7 μM and SI=15, which may provide a useful lead for further molecular optimization.
Co-reporter:Peng Zhan, Xinyong Liu, Yuan Cao, Yan Wang, Christophe Pannecouque, Erik De Clercq
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 20) pp:5368-5371
Publication Date(Web):15 October 2008
DOI:10.1016/j.bmcl.2008.09.055
A novel series of 1,2,3-thiadiazole thioacetanilide (TTA) derivatives have been designed, synthesized and evaluated for its anti-HIV activities in MT-4 cells. Some derivatives proved to be highly effective in inhibiting HIV-1 replication at nanomolar concentrations. Among them, 2-[4-(2,4-dichlorophenyl)-1,2,3-thiadiazol-5-ylthio]-N-(2-nitrophenyl)acetamide 7d2 was identified as the most promising compound (EC50 = 0.059 ± 0.02 μM, CC50 > 283.25 μM, SI > 4883). The structure–activity relationship (SAR) of these novel structural congeners is discussed.A novel series of 1,2,3-thiadiazole thioacetanilide (TTA) derivatives have been designed, synthesized and evaluated for their anti-HIV activities in MT-4 cells. Some derivatives proved to be highly effective in inhibiting HIV-1 replication at nanomolar concentrations. The structure–activity relationship (SAR) is discussed.
Co-reporter:Xian-Chao Cheng, Xin-Yong Liu, Wen-Fang Xu, Xiu-Li Guo, Yang Ou
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 10) pp:3315-3320
Publication Date(Web):15 May 2007
DOI:10.1016/j.bmc.2007.03.033
A series of novel Ligustrazine derivatives was designed, synthesized, and assayed for their protective effects on damaged ECV-304 cells and antiplatelet aggregation activities. The results showed that most Ligustrazine derivatives exhibited lower EC50 values for protective effects on the ECV-304 cells damaged by hydrogen peroxide in comparison with Ligustrazine. And some Ligustrazine derivatives presented better antiplatelet aggregation activities than Ligustrazine. The derivatives containing the bisphenylmethyl pharmacophore (7a–c) exhibited highest potency. Compound 7a displayed most potential protective effects on the ECV-304 cells damaged by hydrogen peroxide, and compound 7c was found to be the most active antiplatelet aggregation agent. Structure–activity relationships were briefly discussed.A series of novel Ligustrazine derivatives was designed and synthesized. Their protective effects on damaged ECV-304 cells and antiplatelet aggregation activities were reported.

![4-[[2-[[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-1,2,4-triazol-3-yl]sulfanyl]acetyl]amino]-3-chlorobenzoic acid](http://img.cochemist.com/ccimg/878700/878670-61-2.png)
![4-[[2-[[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-1,2,4-triazol-3-yl]sulfanyl]acetyl]amino]-3-chlorobenzoic acid](http://img.cochemist.com/ccimg/878700/878670-61-2_b.png)




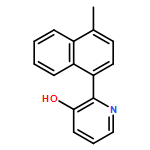
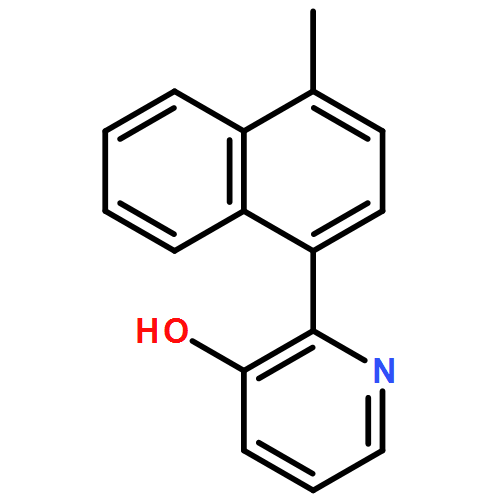
![2-[4-(TRIFLUOROMETHYL)PHENYL]PYRIDIN-3-OL](/data/chemimg/431100/1261886-11-6.png)
![2-[4-(TRIFLUOROMETHYL)PHENYL]PYRIDIN-3-OL](/data/chemimg/431100/1261886-11-6_b.png)
![4-[6-amino-2-(4-cyanoanilino)pyrimidin-4-yl]oxy-3,5-dimethyl-benzonitrile](http://img.cochemist.com/ccimg/939500/939431-68-2.png)
![4-[6-amino-2-(4-cyanoanilino)pyrimidin-4-yl]oxy-3,5-dimethyl-benzonitrile](http://img.cochemist.com/ccimg/939500/939431-68-2_b.png)
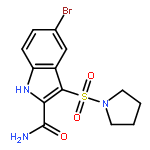
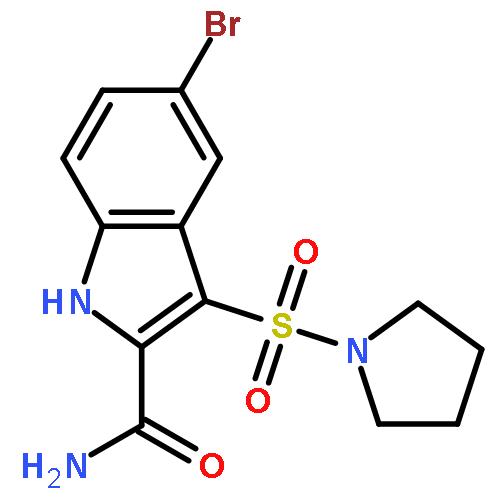
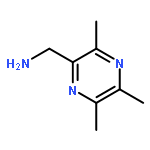
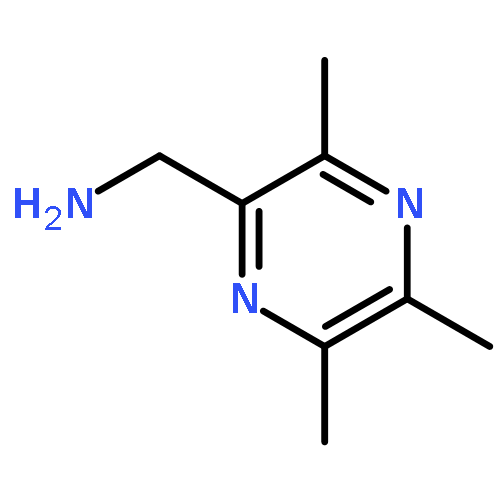
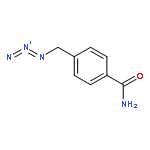
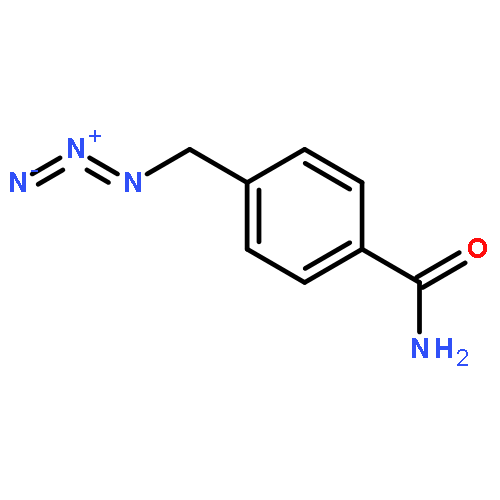


![BENZONITRILE, 4-[(5-CHLORO[1,2,4]TRIAZOLO[1,5-A]PYRIMIDIN-7-YL)AMINO]-](http://img.cochemist.com/ccimg/808800/808733-94-0.png)
![BENZONITRILE, 4-[(5-CHLORO[1,2,4]TRIAZOLO[1,5-A]PYRIMIDIN-7-YL)AMINO]-](http://img.cochemist.com/ccimg/808800/808733-94-0_b.png)





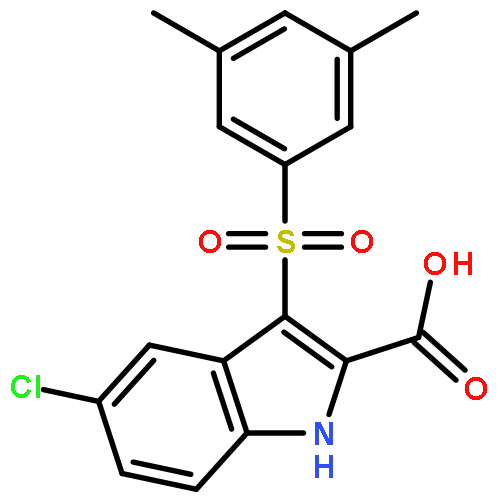

![4-Piperidinamine, 1-[(2,6-dichlorophenyl)methyl]-](http://img.cochemist.com/ccimg/380500/380424-99-7.png)
![4-Piperidinamine, 1-[(2,6-dichlorophenyl)methyl]-](http://img.cochemist.com/ccimg/380500/380424-99-7_b.png)




![Benzonitrile,4-[[4-[(2,4,6-trimethylphenyl)amino]-2-pyrimidinyl]amino]-](http://img.cochemist.com/ccimg/244800/244767-67-7.png)
![Benzonitrile,4-[[4-[(2,4,6-trimethylphenyl)amino]-2-pyrimidinyl]amino]-](http://img.cochemist.com/ccimg/244800/244767-67-7_b.png)
![Ethanone, 1-[4-(azidomethyl)phenyl]-](http://img.cochemist.com/ccimg/223600/223513-47-1.png)
![Ethanone, 1-[4-(azidomethyl)phenyl]-](http://img.cochemist.com/ccimg/223600/223513-47-1_b.png)







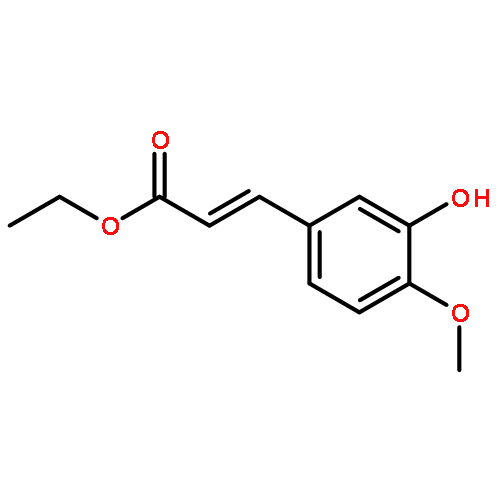






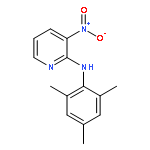





![[1,1':3',1''-Terphenyl]-4'-carboxaldehyde](http://img.cochemist.com/ccimg/136500/136416-26-7.png)
![[1,1':3',1''-Terphenyl]-4'-carboxaldehyde](http://img.cochemist.com/ccimg/136500/136416-26-7_b.png)
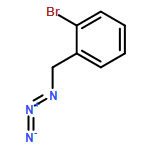

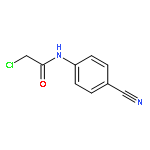
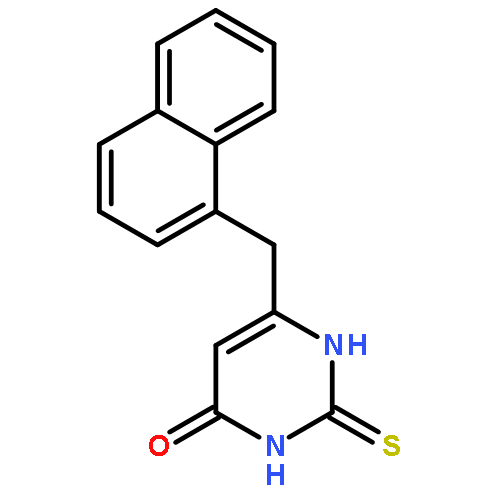
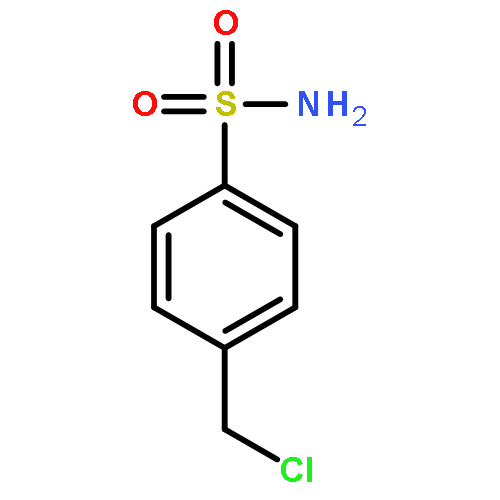
![Benzonitrile, 4-[(4-amino-1-piperidinyl)methyl]-](http://img.cochemist.com/ccimg/92600/92539-21-4.png)
![Benzonitrile, 4-[(4-amino-1-piperidinyl)methyl]-](http://img.cochemist.com/ccimg/92600/92539-21-4_b.png)
![4-Piperidinamine, 1-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/92600/92539-18-9.png)
![4-Piperidinamine, 1-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/92600/92539-18-9_b.png)
![4-Piperidinamine, 1-[(4-bromophenyl)methyl]-](http://img.cochemist.com/ccimg/92600/92539-13-4.png)
![4-Piperidinamine, 1-[(4-bromophenyl)methyl]-](http://img.cochemist.com/ccimg/92600/92539-13-4_b.png)
![4-Piperidinamine, 1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/78500/78471-35-9.png)
![4-Piperidinamine, 1-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/78500/78471-35-9_b.png)


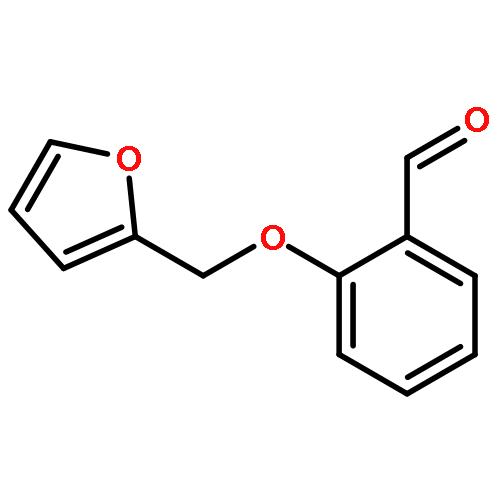

![Glycine, N-[imino(nitroamino)methyl]-](http://img.cochemist.com/ccimg/52500/52498-33-6.png)
![Glycine, N-[imino(nitroamino)methyl]-](http://img.cochemist.com/ccimg/52500/52498-33-6_b.png)


![4-PIPERIDINAMINE, 1-[(2-METHYLPHENYL)METHYL]-](http://img.cochemist.com/ccimg/41300/41220-34-2.png)
![4-PIPERIDINAMINE, 1-[(2-METHYLPHENYL)METHYL]-](http://img.cochemist.com/ccimg/41300/41220-34-2_b.png)






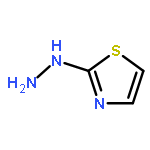
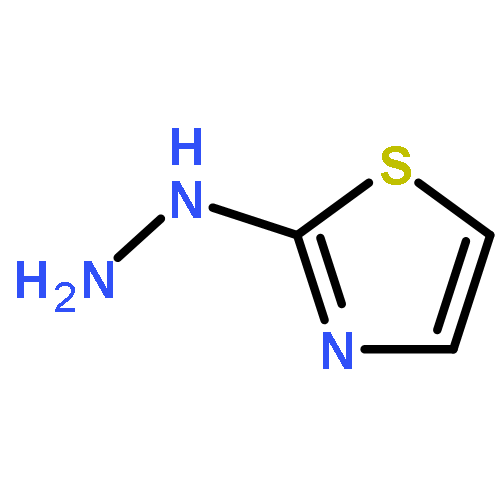




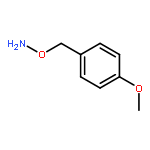
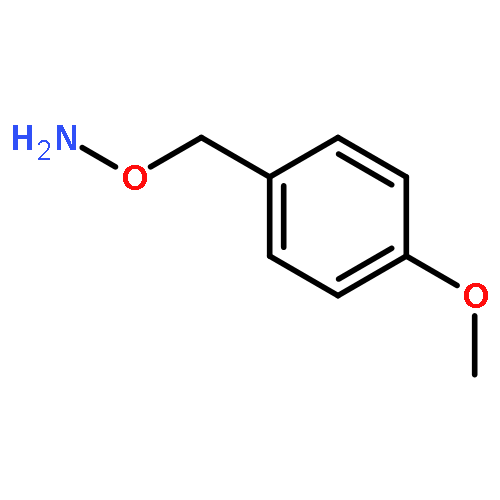



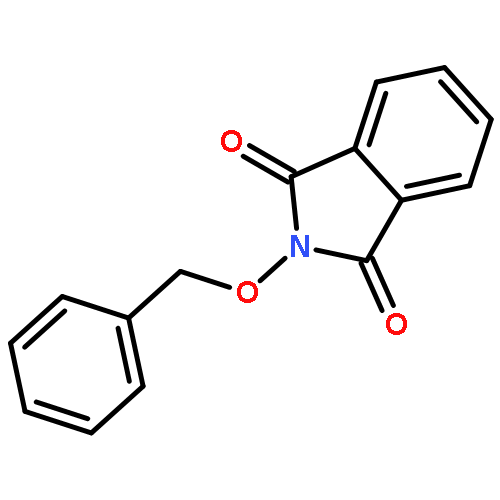

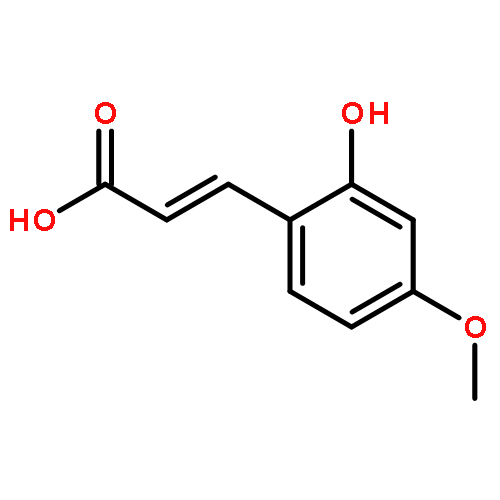


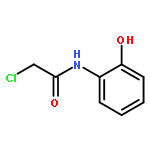
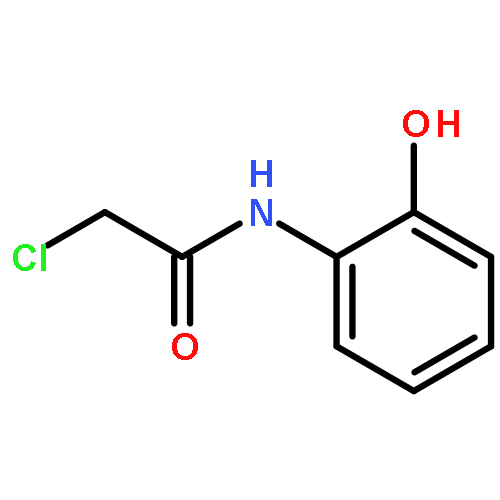


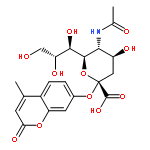
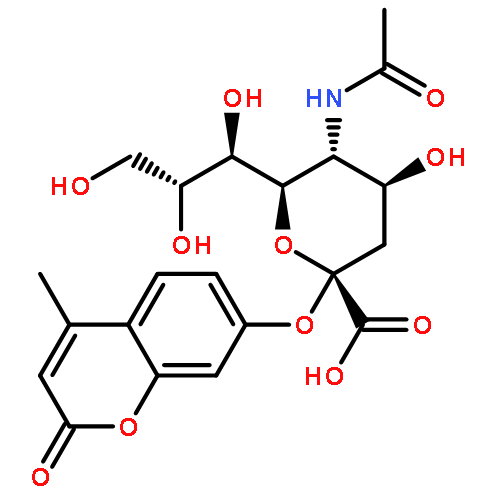















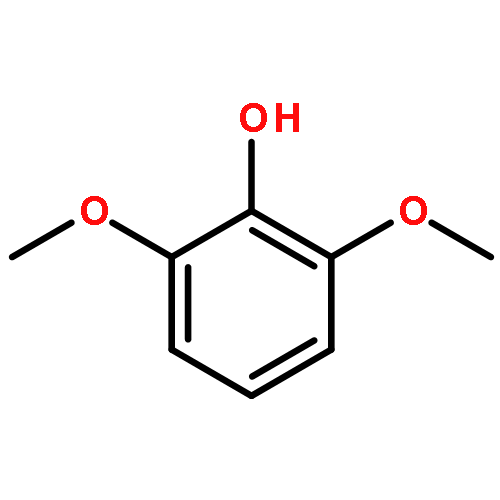
![4H-Dibenzo[de,g]quinoline-10,11-diol,5,6,6a,7-tetrahydro-6-methyl-, (6aR)-](http://img.cochemist.com/ccimg/100/58-00-4.png)
![4H-Dibenzo[de,g]quinoline-10,11-diol,5,6,6a,7-tetrahydro-6-methyl-, (6aR)-](http://img.cochemist.com/ccimg/100/58-00-4_b.png)

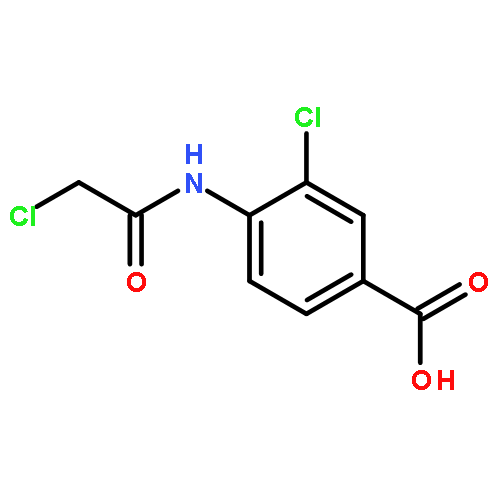
![4-[[4-[4-[(e)-2-cyanoethenyl]-2,6-dimethylanilino]pyrimidin-2-yl]amino]benzonitrile](http://img.cochemist.com/ccimg/500300/500287-72-9.png)
![4-[[4-[4-[(e)-2-cyanoethenyl]-2,6-dimethylanilino]pyrimidin-2-yl]amino]benzonitrile](http://img.cochemist.com/ccimg/500300/500287-72-9_b.png)


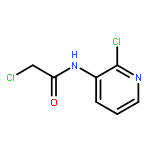
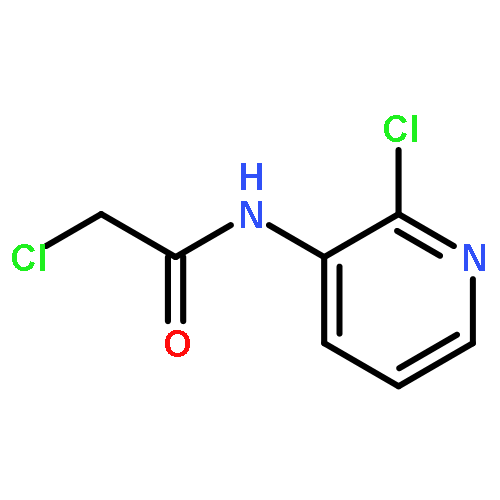
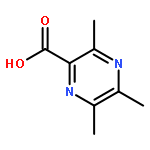
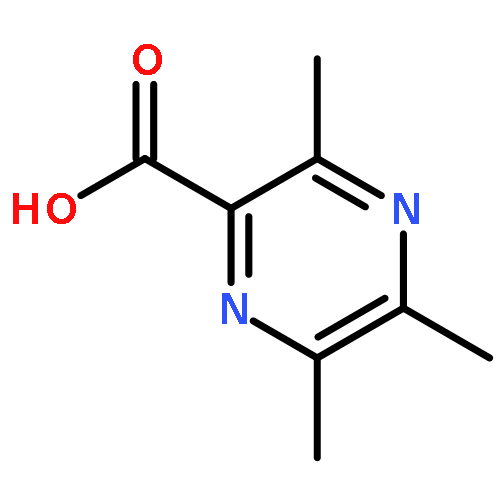
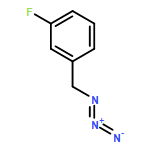
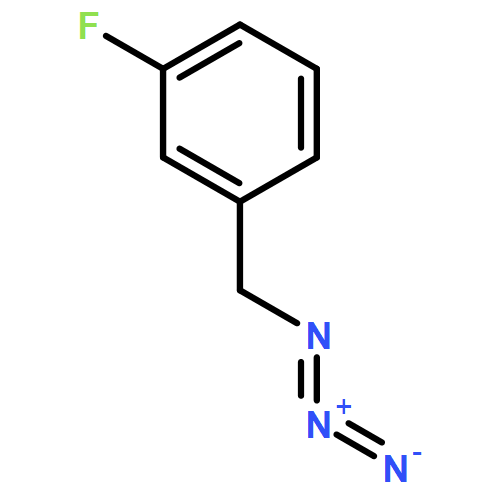

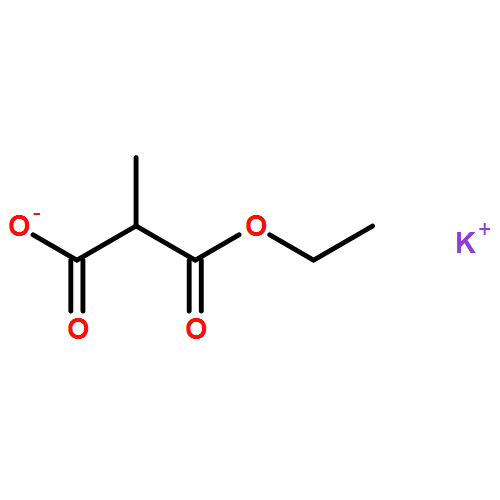
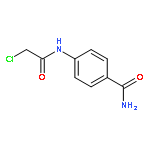

![Benzoic acid, 4-[(2-chloroacetyl)amino]-, methyl ester](/data/chemimg/1518500/82525-64-2.png)
![Benzoic acid, 4-[(2-chloroacetyl)amino]-, methyl ester](/data/chemimg/1518500/82525-64-2_b.png)
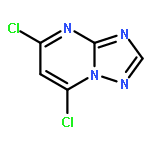
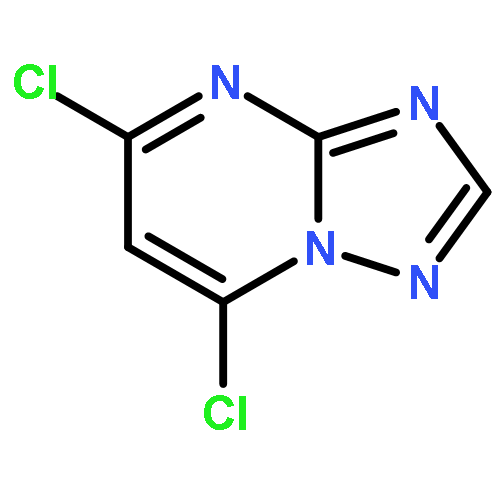
![2-{[(5-CHLORO-2-METHOXYBENZOYL)CARBAMOTHIOYL]AMINO}-3,5-DIIODOBEN<WBR />ZOIC ACID](http://img.cochemist.com/ccimg/72200/72196-97-5.png)
![2-{[(5-CHLORO-2-METHOXYBENZOYL)CARBAMOTHIOYL]AMINO}-3,5-DIIODOBEN<WBR />ZOIC ACID](http://img.cochemist.com/ccimg/72200/72196-97-5_b.png)
![7-HYDROXY-1H-[1,2,4]TRIAZOLO[1,5-A]PYRIMIDIN-5-ONE](http://img.cochemist.com/ccimg/40800/40775-75-5.png)
![7-HYDROXY-1H-[1,2,4]TRIAZOLO[1,5-A]PYRIMIDIN-5-ONE](http://img.cochemist.com/ccimg/40800/40775-75-5_b.png)
![Acetamide,N-[4-(aminosulfonyl)phenyl]-2-chloro-](http://img.cochemist.com/ccimg/15000/14949-01-0.png)
![Acetamide,N-[4-(aminosulfonyl)phenyl]-2-chloro-](http://img.cochemist.com/ccimg/15000/14949-01-0_b.png)
![Pyrazine, [[4-[bis(4-fluorophenyl)methyl]-1-piperazinyl]methyl]trimethyl-](http://img.cochemist.com/ccimg/892400/892396-24-6.png)
![Pyrazine, [[4-[bis(4-fluorophenyl)methyl]-1-piperazinyl]methyl]trimethyl-](http://img.cochemist.com/ccimg/892400/892396-24-6_b.png)









![Pyrazine, [[4-(diphenylmethyl)-1-piperazinyl]methyl]trimethyl-](http://img.cochemist.com/ccimg/892400/892396-23-5.png)
![Pyrazine, [[4-(diphenylmethyl)-1-piperazinyl]methyl]trimethyl-](http://img.cochemist.com/ccimg/892400/892396-23-5_b.png)









