Co-reporter:William Devine, Sarah M. Thomas, Jessey Erath, Kelly A. Bachovchin, Patricia J. Lee, Susan E. Leed, Ana Rodriguez, Richard J. Sciotti, Kojo Mensa-Wilmot, and Michael P. Pollastri
ACS Medicinal Chemistry Letters March 9, 2017 Volume 8(Issue 3) pp:
Publication Date(Web):February 5, 2017
DOI:10.1021/acsmedchemlett.7b00011
Human African trypanosomiasis (HAT), Chagas disease, and leishmaniasis present a significant burden across the developing world. Existing therapeutics for these protozoal neglected tropical diseases suffer from severe side effects and toxicity. Previously, NEU-1045 (3) was identified as a promising lead with cross-pathogen activity, though it possessed poor physicochemical properties. We have designed a library of analogues with improved calculated physicochemical properties built on the quinoline scaffold of 3 incorporating small, polar aminoheterocycles in place of the 4-(3-fluorobenzyloxy)aniline substituent. We report the biological activity of these inhibitors against Trypanosoma brucei (HAT), T. cruzi (Chagas disease), and Leishmania major (cutaneous leishmaniasis) and describe the identification of N-(5-chloropyrimidin-2-yl)-6-(4-(morpholinosulfonyl)phenyl)quinolin-4-amine (13t) as a promising inhibitor of L. major proliferation and 6-(4-(morpholinosulfonyl)phenyl)-N-(pyrimidin-4-yl)quinolin-4-amine (13j), a potent inhibitor of T. brucei proliferation with improved drug-like properties.Keywords: Antiparasitic agents; Chagas disease; human African trypanosomiasis; Leishmania major; leishmaniasis; Plasmodium falciparum; Trypanosoma brucei; Trypanosoma cruzi;
Co-reporter:William G. Devine, Rosario Diaz-Gonzalez, Gloria Ceballos-Perez, Domingo Rojas, Takashi Satoh, Westley Tear, Ranae M. Ranade, Ximena Barros-Álvarez, Wim G. J. Hol, Frederick S. Buckner, Miguel Navarro, and Michael P. Pollastri
ACS Infectious Diseases 2017 Volume 3(Issue 3) pp:
Publication Date(Web):January 22, 2017
DOI:10.1021/acsinfecdis.6b00202
Human African trypanosomiasis is a neglected tropical disease that is lethal if left untreated. Existing therapeutics have limited efficacy and severe associated toxicities. 2-(2-(((3-((1H-Benzo[d]imidazol-2-yl)amino)propyl)amino)methyl)-4,6-dichloro-1H-indol-1-yl)ethan-1-ol (NEU-1053) has recently been identified from a high-throughput screen of >42,000 compounds as a highly potent and fast-acting trypanocidal agent capable of curing a bloodstream infection of Trypanosoma brucei in mice. We have designed a library of analogues to probe the structure–activity relationship and improve the predicted central nervous system (CNS) exposure of NEU-1053. We report the activity of these inhibitors of T. brucei, the efficacy of NEU-1053 in a murine CNS model of infection, and identification of the target of NEU-1053 via X-ray crystallography.Keywords: medicinal chemistry; methionyl-tRNA synthetase; Trypanosoma brucei;
Co-reporter:Jennifer L. Woodring, Kelly A. Bachovchin, Kimberly G. Brady, Mitchell F. Gallerstein, Jessey Erath, Scott Tanghe, Susan E. Leed, Ana Rodriguez, Kojo Mensa-Wilmot, Richard J. Sciotti, Michael P. Pollastri
European Journal of Medicinal Chemistry 2017 Volume 141(Volume 141) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.ejmech.2017.10.007
•4-Anilinoquinazoline-derived inhibitors of Trypanosoma brucei proliferation were redesigned for improved properties, such as LogP, LLE, MPO score.•Replacement of the large, lipophililc headgroup with small aromatic amines provided compounds with improvements in properties.•Various bioisosteric replacements were made for phenyl ring in tail region of molecule.•All compounds were tested against other protozoan pathogens, such a Trypanosoma cruzi, Leishmania major, and Plasmodium falciparum.Human African trypanosomiasis (HAT) is a deadly disease in need of new chemotherapeutics that can cross into the central nervous system. We previously reported the discovery of 2 (NEU-617), a small molecule with activity against T. brucei bloodstream proliferation. Further optimization of 2 to improve the physicochemical properties (LogP, LLE, [1], and MPO score) [2] have led us to twelve sub-micromolar compounds, most importantly the headgroup variants 9i and 9j, and the linker variant 18. Although these 3 compounds had reduced potency compared to 2, they all had improved LogP, LLE and MPO scores. Cross-screening these analogs against other protozoan parasites uncovered 9o with potent activity towards T. brucei, T. cruzi and L. major, while four others compounds (17, 18, 21, 26) showed activity towards P. falciparum D6. This reinforces the effectiveness of lead repurposing for the discovery of new protozoan disease therapeutics.Download high-res image (136KB)Download full-size image
Co-reporter:Dana M. Klug, Michael H. Gelb, Michael P. Pollastri
Bioorganic & Medicinal Chemistry Letters 2016 26(11) pp: 2569-2576
Publication Date(Web):1 June 2016
DOI:10.1016/j.bmcl.2016.03.103
Neglected tropical diseases (NTDs) and other diseases of the developing world, such as malaria, attract research investments that are disproportionately low compared to their impact on human health worldwide. Therefore, pragmatic methods for launching new drug discovery programs have emerged that repurpose existing chemical matter as new drugs or new starting points for optimization. In this Digest we describe applications of different repurposing approaches for NTDs, and provide a means by which these approaches may be differentiated from each other. These include drug repurposing, target repurposing, target class repurposing, and lead repurposing.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Emanuele Amata, Hualin Xi, Gonzalo Colmenarejo, Rosario Gonzalez-Diaz, Carlos Cordon-Obras, Manuela Berlanga, Pilar Manzano, Jessey Erath, Norma E. Roncal, Patricia J. Lee, Susan E. Leed, Ana Rodriguez, Richard J. Sciotti, Miguel Navarro, and Michael P. Pollastri
ACS Infectious Diseases 2016 Volume 2(Issue 3) pp:180
Publication Date(Web):January 17, 2016
DOI:10.1021/acsinfecdis.5b00136
A kinase-targeting cell-based high-throughput screen (HTS) against Trypanosoma brucei was recently reported, and this screening set included the Published Kinase Inhibitor Set (PKIS). From the PKIS was identified 53 compounds with pEC50 ≥ 6. Utilizing the published data available for the PKIS, a statistical analysis of these active antiparasitic compounds was performed, allowing identification of a set of human kinases having inhibitors that show a high likelihood for blocking T. brucei cellular proliferation in vitro. This observation was confirmed by testing other established inhibitors of these human kinases and by mining past screening campaigns at GlaxoSmithKline. Overall, although the parasite targets of action are not known, inhibitors of this set of human kinases displayed an enhanced hit rate relative to a random kinase-targeting HTS campaign, suggesting that repurposing efforts should focus primarily on inhibitors of these specific human kinases. We therefore term this statistical analysis-driven approach “preferred lead repurposing”.Keywords: Leishmania major; Plasmodium falciparum; preferred lead repurposing; Published Kinase Inhibitor Set; Trypanosoma brucei; Trypanosoma cruzi
Co-reporter:William Devine; Jennifer L. Woodring; Uma Swaminathan; Emanuele Amata; Gautam Patel; Jessey Erath; Norma E. Roncal; Patricia J. Lee; Susan E. Leed; Ana Rodriguez; Kojo Mensa-Wilmot; Richard J. Sciotti;Michael P. Pollastri
Journal of Medicinal Chemistry 2015 Volume 58(Issue 14) pp:5522-5537
Publication Date(Web):June 18, 2015
DOI:10.1021/acs.jmedchem.5b00515
Tropical protozoal infections are a significant cause of morbidity and mortality worldwide; four in particular (human African trypanosomiasis (HAT), Chagas disease, cutaneous leishmaniasis, and malaria) have an estimated combined burden of over 87 million disability-adjusted life years. New drugs are needed for each of these diseases. Building on the previous identification of NEU-617 (1) as a potent and nontoxic inhibitor of proliferation for the HAT pathogen (Trypanosoma brucei), we have now tested this class of analogs against other protozoal species: T. cruzi (Chagas disease), Leishmania major (cutaneous leishmaniasis), and Plasmodium falciparum (malaria). Based on hits identified in this screening campaign, we describe the preparation of several replacements for the quinazoline scaffold and report these inhibitors’ biological activities against these parasites. In doing this, we have identified several potent proliferation inhibitors for each pathogen, such as 4-((3-chloro-4-((3-fluorobenzyl)oxy)phenyl)amino)-6-(4-((4-methyl-1,4-diazepan-1-yl)sulfonyl)phenyl)quinoline-3-carbonitrile (NEU-924, 83) for T. cruzi and N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-7-(4-((4-methyl-1,4-diazepan-1-yl)sulfonyl)phenyl)cinnolin-4-amine (NEU-1017, 68) for L. major and P. falciparum.
Co-reporter:Stefan O. Ochiana;Nicholas D. Bl;Luca Settimo;Robert K. Campbell;Michael P. Pollastri
Chemical Biology & Drug Design 2015 Volume 85( Issue 5) pp:549-564
Publication Date(Web):
DOI:10.1111/cbdd.12443
Cyclic nucleotide phosphodiesterases (PDEs) have been identified as important enzyme targets for drug development in both humans and Trypanosoma brucei, the causative agent of human African trypanosomiasis. With this in mind, we recently reported the profiling of a range of human phosphodiesterase inhibitors, showing that human PDE4 inhibitors tend to display the best potency against the trypanosomal phosphodiesterase TbrPDEB1. Among these was GSK-256066, a potent inhibitor of human PDE4 and a weak inhibitor of TbrPDEB1. In this report, we describe the results of a structure–activity relationship study of this chemotype, leading to the discovery of analogs with improved potency against TbrPDEB1 and micromolar inhibition of T. brucei cellular growth. We rationalize the potency trends via molecular docking of the new inhibitors into a recently reported apo structure of TbrPDEB1. The studies in this article will inform future efforts in repurposing human PDE inhibitors as antitrypanosomal agents.
Co-reporter:Emanuele Amata, Nicholas D. Bland, Robert K. Campbell, Michael P. Pollastri
Tetrahedron Letters 2015 Volume 56(Issue 21) pp:2832-2835
Publication Date(Web):20 May 2015
DOI:10.1016/j.tetlet.2015.04.061
Human African trypanosomiasis (HAT) is a parasitic disease, caused by the protozoan pathogen Trypanosoma brucei, which affects thousands every year and which is in need of new therapeutics. Herein we report the synthesis and assessment of a series of pyrrolidine and pyrazolone derivatives of human phosphodiesterase 4 (hPDE4) inhibitors for the assessment of their activity against the trypanosomal phosphodiesterase TbrPDEB1. The synthesized compounds showed weak potency against TbrPDEB1.
Co-reporter:Jennifer L. Woodring, Gautam Patel, Jessey Erath, Ranjan Behera, Patricia J. Lee, Susan E. Leed, Ana Rodriguez, Richard J. Sciotti, Kojo Mensa-Wilmot and Michael P. Pollastri
MedChemComm 2015 vol. 6(Issue 2) pp:339-346
Publication Date(Web):14 Nov 2014
DOI:10.1039/C4MD00441H
Target repurposing is a proven method for finding new lead compounds that target Trypanosoma brucei, the causative agent of human African trypanosomiasis. Due to the recent discovery of a lapatinib-derived analog 2 with excellent potency against T. brucei (EC50 = 42 nM) and selectivity over human host cells, we have explored other classes of human tyrosine kinase inhibitor scaffolds in order to expand the range of chemotypes for pursuit. Following library expansion, we found compound 11e to have an EC50 of 84 nM against T. brucei cells while maintaining selectivity over human hepatocytes. In addition, the library was tested against causative agents of Chagas' disease, leishmaniasis, and malaria. Two analogs with sub-micromolar potencies for T. cruzi (4j) and Plasmodium falciparum (11j) were discovered, along with an analog with considerable potency against Leishmania major amastigotes (4e). Besides identifying new and potent protozoan growth inhibitors, these data highlight the value of concurrent screening of a chemical library against different protozoan parasites.
Co-reporter:Christopher Merritt, Lisseth E. Silva, Angela L. Tanner, Kenneth Stuart, and Michael P. Pollastri
Chemical Reviews 2014 Volume 114(Issue 22) pp:11280
Publication Date(Web):October 7, 2014
DOI:10.1021/cr500197d
Co-reporter:João D. Seixas ; Sandra A. Luengo-Arratta ; Rosario Diaz ; Manuel Saldivia ; Domingo I. Rojas-Barros ; Pilar Manzano ; Silvia Gonzalez ; Manuela Berlanga ; Terry K. Smith ; Miguel Navarro ;Michael P. Pollastri
Journal of Medicinal Chemistry 2014 Volume 57(Issue 11) pp:4834-4848
Publication Date(Web):May 7, 2014
DOI:10.1021/jm500361r
Compound NVP-BEZ235 (1) is a potent inhibitor of human phospoinositide-3-kinases and mammalian target of rapamycin (mTOR) that also showed high inhibitory potency against Trypanosoma brucei cultures. With an eye toward using 1 as a starting point for anti-trypanosomal drug discovery, we report efforts to reduce host cell toxicity, to improve the physicochemical properties, and to improve the selectivity profile over human kinases. In this work, we have developed structure–activity relationships for analogues of 1 and have prepared analogues of 1 with improved solubility properties and good predicted central nervous system exposure. In this way, we have identified 4e, 9, 16e, and 16g as the most promising leads to date. We also report cell phenotype and phospholipidomic studies that suggest that these compounds exert their anti-trypanosomal effects, at least in part, by inhibition of lipid kinases.
Co-reporter:Gautam Patel, Norma E. Roncal, Patricia J. Lee, Susan E. Leed, Jessey Erath, Ana Rodriguez, Richard J. Sciotti and Michael P. Pollastri
MedChemComm 2014 vol. 5(Issue 5) pp:655-658
Publication Date(Web):17 Mar 2014
DOI:10.1039/C4MD00045E
Hesperadin, an established human Aurora B inhibitor, was tested against cultures of Trypanosoma brucei, Leishmania major, and Plasmodium falciparum, and was identified to be a potent proliferation inhibitor. A series of analogs was designed and tested to establish the initial structure–activity relationships for each parasite. In this study, we identified multiple non-toxic compounds with high potency against T. brucei and P. falciparum with good selectivity. These compounds may represent an opportunity for continued optimization.
Co-reporter:Grasiella Andriani ; Emanuele Amata ; Joel Beatty ; Zeke Clements ; Brian J. Coffey ; Gilles Courtemanche ; William Devine ; Jessey Erath ; Cristin E. Juda ; Zdzislaw Wawrzak ; JodiAnne T. Wood ; Galina I. Lepesheva ; Ana Rodriguez ;Michael P. Pollastri
Journal of Medicinal Chemistry 2013 Volume 56(Issue 6) pp:2556-2567
Publication Date(Web):February 28, 2013
DOI:10.1021/jm400012e
Chagas disease is caused by the intracellular protozoan parasite Trypanosomal cruzi, and current drugs are lacking in terms of desired safety and efficacy profiles. Following on a recently reported high-throughput screening campaign, we have explored initial structure–activity relationships around a class of imidazole-based compounds. This profiling has uncovered compounds 4c (NEU321) and 4j (NEU704), which are potent against in vitro cultures of T. cruzi and are greater than 160-fold selective over host cells. We report in vitro drug metabolism and properties profiling of 4c and show that this chemotype inhibits the T. cruzi CYP51 enzyme, an observation confirmed by X-ray crystallographic analysis. We compare the binding orientation of 4c to that of other, previously reported inhibitors. We show that 4c displays a significantly better ligand efficiency and a shorter synthetic route over previously disclosed CYP51 inhibitors, and should therefore be considered a promising lead compound for further optimization.
Co-reporter:Gautam Patel ; Caitlin E. Karver ; Ranjan Behera ; Paul J. Guyett ; Catherine Sullenberger ; Peter Edwards ; Norma E. Roncal ; Kojo Mensa-Wilmot ;Michael P. Pollastri
Journal of Medicinal Chemistry 2013 Volume 56(Issue 10) pp:3820-3832
Publication Date(Web):April 18, 2013
DOI:10.1021/jm400349k
Human African trypanosomiasis (HAT) is a neglected tropical disease caused by the protozoan parasite Trypanosoma brucei. Because drugs in use against HAT are toxic and require intravenous dosing, new drugs are needed. Initiating lead discovery campaigns by using chemical scaffolds from drugs approved for other indications can speed up drug discovery for neglected diseases. We demonstrated recently that the 4-anilinoquinazolines lapatinib (GW572016, 1) and canertinib (CI-1033) kill T. brucei with low micromolar EC50 values. We now report promising activity of analogues of 1, which provided an excellent starting point for optimization of the chemotype. Our compound optimization that has led to synthesis of several potent 4-anilinoquinazolines, including NEU617, 23a, a highly potent, orally bioavailable inhibitor of trypanosome replication. At the cellular level, 23a blocks duplication of the kinetoplast and arrests cytokinesis, making it a new chemical tool for studying regulation of the trypanosome cell cycle.
Co-reporter:Stefan O. Ochiana, Vidya Pandarinath, Zhouxi Wang, Rishika Kapoor, Mary Jo Ondrechen, Larry Ruben, Michael P. Pollastri
European Journal of Medicinal Chemistry 2013 Volume 62() pp:777-784
Publication Date(Web):April 2013
DOI:10.1016/j.ejmech.2012.07.038
New drugs for neglected tropical diseases such as human African trypanosomiasis (HAT) are needed, yet drug discovery efforts are not often focused on this area due to cost. Target repurposing, achieved by the matching of essential parasite enzymes to those human enzymes that have been successfully inhibited by small molecule drugs, provides an attractive means by which new drug optimization programs can be pragmatically initiated. In this report we describe our results in repurposing an established class of human Aurora kinase inhibitors, typified by danusertib (1), which we have observed to be an inhibitor of trypanosomal Aurora kinase 1 (TbAUK1) and effective in parasite killing in vitro. Informed by homology modeling and docking, a series of analogs of 1 were prepared that explored the scope of the chemotype and provided a nearly 25-fold improvement in cellular selectivity for parasite cells over human cells.Graphical abstractHighlights► We assessed a class of human Aurora kinase inhibitors against Trypanosoma brucei. ► The human Aurora inhibitor danusertib inhibits TbAUK1 and T. brucei growth. ► Analogs of danusertib were designed based on a TbAUK1 homology model. ► New analogs show improved selectivity for killing T. brucei cells over host cells. ► We show that target repurposing can provide a rapid start for NTD drug discovery.
Co-reporter:Jennifer L. Woodring, Nicholas D. Bland, Stefan O. Ochiana, Robert K. Campbell, Michael P. Pollastri
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 21) pp:5971-5974
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmcl.2013.08.057
Human African trypanosomiasis (HAT) is a parasitic neglected tropical disease that affects 10,000 patients each year. Current treatments are sub-optimal, and the disease is fatal if not treated. Herein, we report our continuing efforts to repurpose the human phosphodiesterase 4 (hPDE4) inhibitor piclamilast to target trypanosomal phosphodiesterase TbrPDEB1. We prepared a range of substituted heterocyclic replacements for the 4-amino-3,5-dichloro-pyridine headgroup of piclamilast, and found that these compounds exhibited weak inhibitory activity of TbrPDEB1.
Co-reporter:Cuihua Wang, Trent D. Ashton, Alden Gustafson, Nicholas D. Bland, Stefan O. Ochiana, Robert K. Campbell, Michael P. Pollastri
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 7) pp:2579-2581
Publication Date(Web):1 April 2012
DOI:10.1016/j.bmcl.2012.01.119
Parasitic diseases, such as African sleeping sickness, have a significant impact on the health and well-being in the poorest regions of the world. Pragmatic drug discovery efforts are needed to find new therapeutic agents. In this Letter we describe target repurposing efforts focused on trypanosomal phosphodiesterases. We outline the synthesis and biological evaluation of analogs of sildenafil (1), a human PDE5 inhibitor, for activities against trypanosomal PDEB1 (TbrPDEB1). We find that, while low potency analogs can be prepared, this chemical class is a sub-optimal starting point for further development of TbrPDE inhibitors.
Co-reporter:Stefan O. Ochiana, Alden Gustafson, Nicholas D. Bland, Cuihua Wang, Michael J. Russo, Robert K. Campbell, Michael P. Pollastri
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 7) pp:2582-2584
Publication Date(Web):1 April 2012
DOI:10.1016/j.bmcl.2012.01.118
In this Letter we describe our ongoing target repurposing efforts focused on discovery of inhibitors of the essential trypanosomal phosphodiesterase TbrPDEB1. This enzyme has been implicated in virulence of Trypanosoma brucei, the causative agent of human African trypanosomiasis (HAT). We outline the synthesis and biological evaluation of analogs of tadalafil, a human PDE5 inhibitor currently utilized for treatment of erectile dysfunction, and report that these analogs are weak inhibitors of TbrPDEB1.
Co-reporter:Nicholas D. Bland ; Cuihua Wang ; Craig Tallman ; Alden E. Gustafson ; Zhouxi Wang ; Trent D. Ashton ; Stefan O. Ochiana ; Gregory McAllister ; Kristina Cotter ; Anna P. Fang ; Lara Gechijian ; Norman Garceau ; Rajiv Gangurde ; Ron Ortenberg ; Mary Jo Ondrechen ; Robert K. Campbell ;Michael P. Pollastri
Journal of Medicinal Chemistry 2011 Volume 54(Issue 23) pp:8188-8194
Publication Date(Web):October 24, 2011
DOI:10.1021/jm201148s
Neglected tropical disease drug discovery requires application of pragmatic and efficient methods for development of new therapeutic agents. In this report, we describe our target repurposing efforts for the essential phosphodiesterase (PDE) enzymes TbrPDEB1 and TbrPDEB2 of Trypanosoma brucei, the causative agent for human African trypanosomiasis (HAT). We describe protein expression and purification, assay development, and benchmark screening of a collection of 20 established human PDE inhibitors. We disclose that the human PDE4 inhibitor piclamilast, and some of its analogues, show modest inhibition of TbrPDEB1 and B2 and quickly kill the bloodstream form of the subspecies T. brucei brucei. We also report the development of a homology model of TbrPDEB1 that is useful for understanding the compound–enzyme interactions and for comparing the parasitic and human enzymes. Our profiling and early medicinal chemistry results strongly suggest that human PDE4 chemotypes represent a better starting point for optimization of TbrPDEB inhibitors than those that target any other human PDEs.





![Diethyl {[(4-iodo-2-methylphenyl)amino]methylene}malonate](http://img.cochemist.com/ccimg/951100/951006-38-5.png)
![Diethyl {[(4-iodo-2-methylphenyl)amino]methylene}malonate](http://img.cochemist.com/ccimg/951100/951006-38-5_b.png)
![2-Methyl-2-(4-(3-methyl-2-oxo-8-(quinolin-3-yl)-2,3-dihydro-1H-imidazo[4,5-c]quinolin-1-yl)phenyl)propanenitrile](http://img.cochemist.com/ccimg/915100/915019-65-7.png)
![2-Methyl-2-(4-(3-methyl-2-oxo-8-(quinolin-3-yl)-2,3-dihydro-1H-imidazo[4,5-c]quinolin-1-yl)phenyl)propanenitrile](http://img.cochemist.com/ccimg/915100/915019-65-7_b.png)
![1-methyl-4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfonylpiperazine](http://img.cochemist.com/ccimg/914700/914610-39-2.png)
![1-methyl-4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfonylpiperazine](http://img.cochemist.com/ccimg/914700/914610-39-2_b.png)
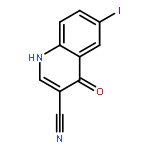
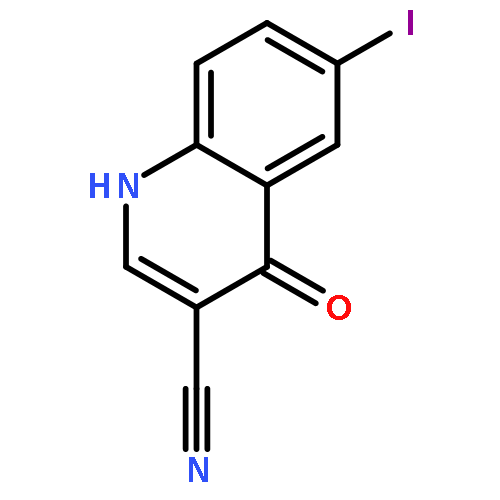
![2-Propenoic acid, 2-cyano-3-[(4-iodophenyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/872600/872576-90-4.png)
![2-Propenoic acid, 2-cyano-3-[(4-iodophenyl)amino]-, ethyl ester, (2Z)-](http://img.cochemist.com/ccimg/872600/872576-90-4_b.png)
![3-[8-amino-1-(2-phenylquinolin-7-yl)imidazo[1,5-a]pyrazin-3-yl]-1-methylcyclobutan-1-ol](http://img.cochemist.com/ccimg/867200/867160-71-2.png)
![3-[8-amino-1-(2-phenylquinolin-7-yl)imidazo[1,5-a]pyrazin-3-yl]-1-methylcyclobutan-1-ol](http://img.cochemist.com/ccimg/867200/867160-71-2_b.png)
![4-Benzyl-2-(naphthalen-1-yl)-[1,2,4]thiadiazolidine-3,5-dione](http://img.cochemist.com/ccimg/865900/865854-05-3.png)
![4-Benzyl-2-(naphthalen-1-yl)-[1,2,4]thiadiazolidine-3,5-dione](http://img.cochemist.com/ccimg/865900/865854-05-3_b.png)
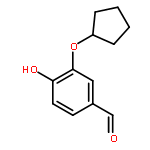
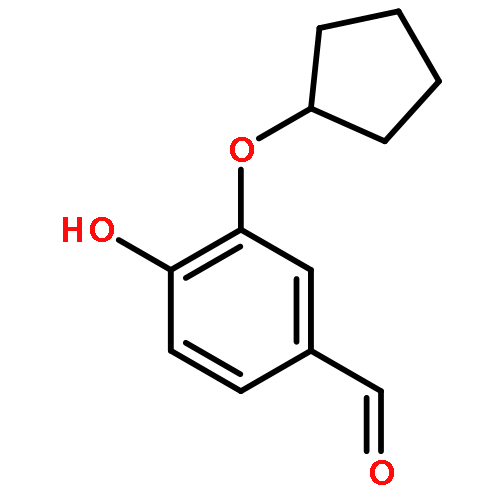
![2-methyl-2-[4-[3-methyl-2-oxo-8-(2-pyridin-3-ylethynyl)imidazo[4,5-c]quinolin-1-yl]phenyl]propanenitrile](http://img.cochemist.com/ccimg/854000/853910-61-9.png)
![2-methyl-2-[4-[3-methyl-2-oxo-8-(2-pyridin-3-ylethynyl)imidazo[4,5-c]quinolin-1-yl]phenyl]propanenitrile](http://img.cochemist.com/ccimg/854000/853910-61-9_b.png)
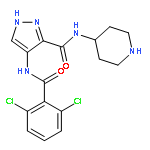
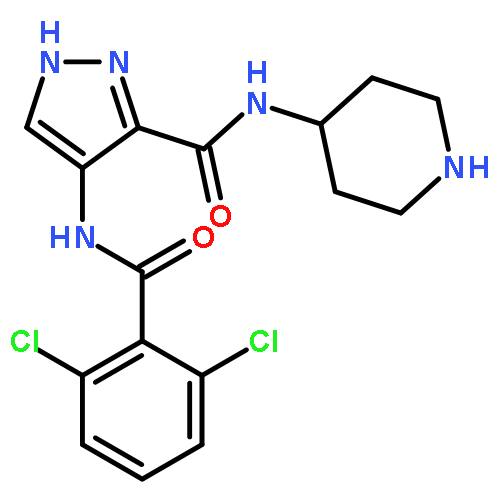
![N-[5-[(2r)-2-methoxy-2-phenylacetyl]-4,6-dihydro-1h-pyrrolo[3,4-c]pyrazol-3-yl]-4-(4-methylpiperazin-1-yl)benzamide](http://img.cochemist.com/ccimg/827400/827318-97-8.png)
![N-[5-[(2r)-2-methoxy-2-phenylacetyl]-4,6-dihydro-1h-pyrrolo[3,4-c]pyrazol-3-yl]-4-(4-methylpiperazin-1-yl)benzamide](http://img.cochemist.com/ccimg/827400/827318-97-8_b.png)
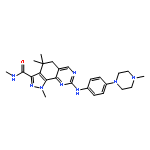
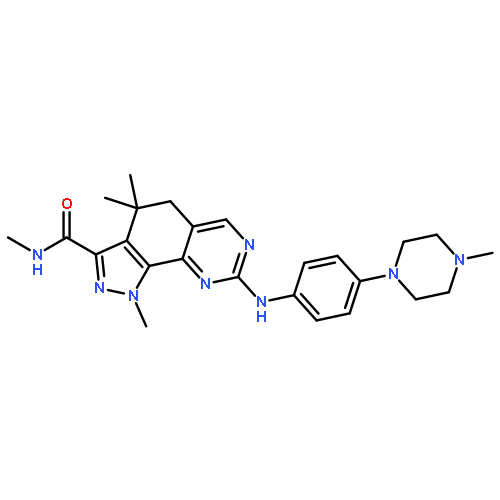
![Methyl 3-({3-carbamoyl-4-[(3-methoxyphenyl)amino]-8-methyl-6-quin olinyl}sulfanyl)benzoate](http://img.cochemist.com/ccimg/801400/801316-10-9.png)
![Methyl 3-({3-carbamoyl-4-[(3-methoxyphenyl)amino]-8-methyl-6-quin olinyl}sulfanyl)benzoate](http://img.cochemist.com/ccimg/801400/801316-10-9_b.png)
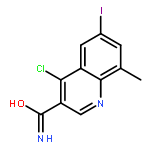
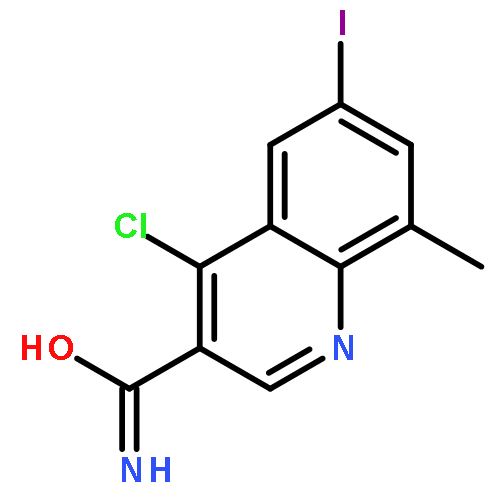
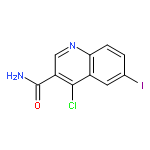
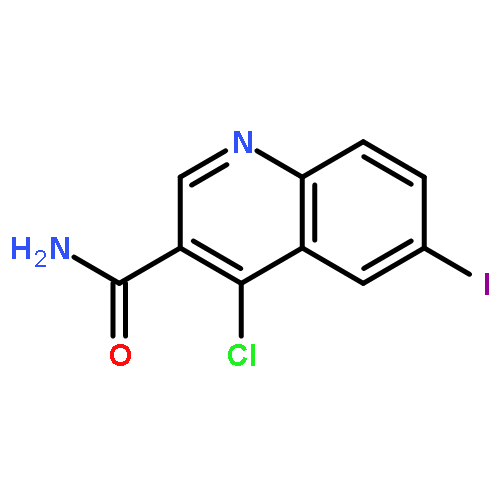
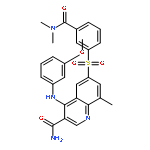
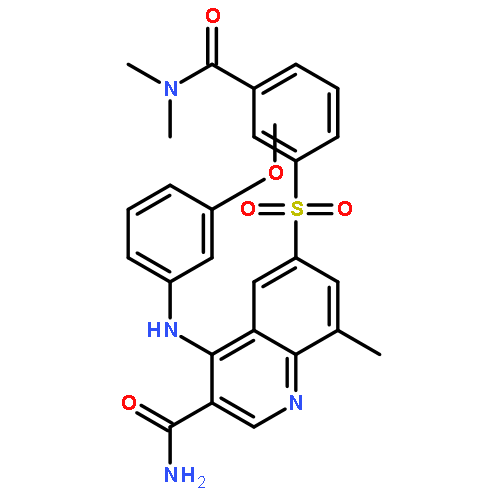
![Benzoic acid,
3-[[3-(aminocarbonyl)-4-[(3-methoxyphenyl)amino]-8-methyl-6-quinolinyl
]sulfonyl]-](http://img.cochemist.com/ccimg/801400/801311-41-1.png)
![Benzoic acid,
3-[[3-(aminocarbonyl)-4-[(3-methoxyphenyl)amino]-8-methyl-6-quinolinyl
]sulfonyl]-](http://img.cochemist.com/ccimg/801400/801311-41-1_b.png)
![Benzoic acid,
3-[[3-(aminocarbonyl)-4-[(3-methoxyphenyl)amino]-8-methyl-6-quinolinyl
]sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/801400/801311-24-0.png)
![Benzoic acid,
3-[[3-(aminocarbonyl)-4-[(3-methoxyphenyl)amino]-8-methyl-6-quinolinyl
]sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/801400/801311-24-0_b.png)


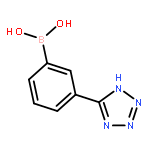
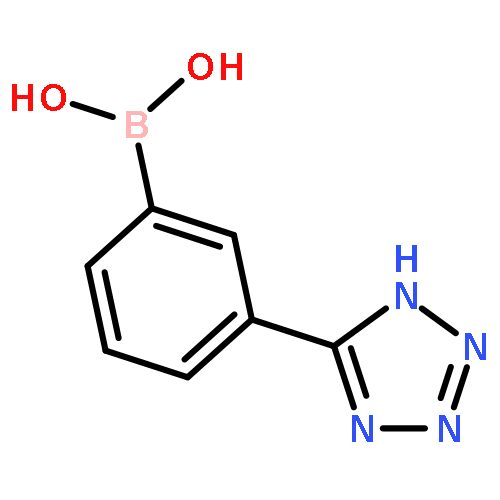
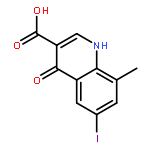
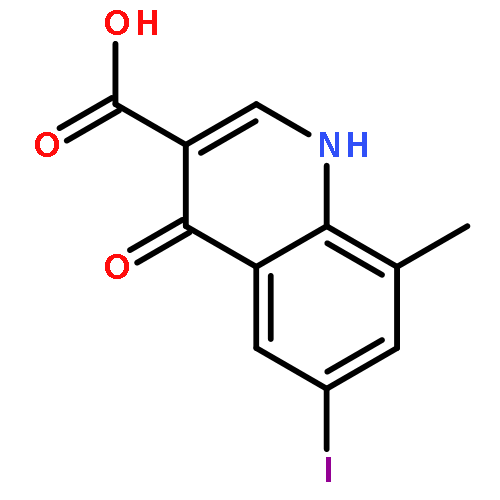
![5-Chloro-N2-[2-methoxy-4-[4-(4-methyl-1-piperazinyl)-1-piperidinyl]phenyl]-N4-[2-[(1-methylethyl)sulfonyl]phenyl]-2,4-pyrimidinediamine](http://img.cochemist.com/ccimg/761500/761439-42-3.png)
![5-Chloro-N2-[2-methoxy-4-[4-(4-methyl-1-piperazinyl)-1-piperidinyl]phenyl]-N4-[2-[(1-methylethyl)sulfonyl]phenyl]-2,4-pyrimidinediamine](http://img.cochemist.com/ccimg/761500/761439-42-3_b.png)






![4-Piperidinecarbonitrile, 4-[3-(cyclopentyloxy)-4-methoxyphenyl]-](http://img.cochemist.com/ccimg/666200/666179-73-3.png)
![4-Piperidinecarbonitrile, 4-[3-(cyclopentyloxy)-4-methoxyphenyl]-](http://img.cochemist.com/ccimg/666200/666179-73-3_b.png)
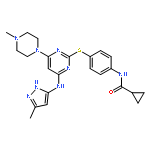





![Glycine, N-[2-methoxy-5-[[[(1E)-2-(2,4,6-trimethoxyphenyl)ethenyl]sulfonyl]methyl]phenyl]-](http://img.cochemist.com/ccimg/592600/592542-59-1.png)
![Glycine, N-[2-methoxy-5-[[[(1E)-2-(2,4,6-trimethoxyphenyl)ethenyl]sulfonyl]methyl]phenyl]-](http://img.cochemist.com/ccimg/592600/592542-59-1_b.png)

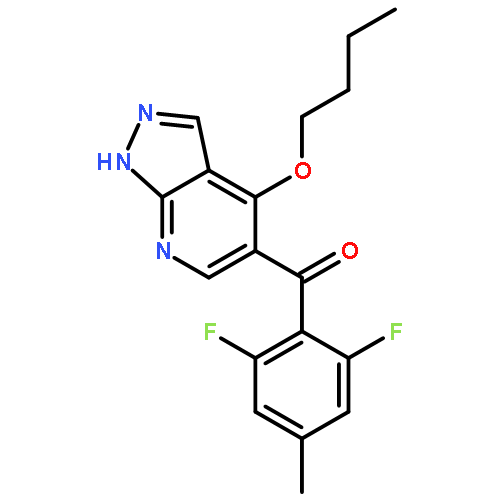



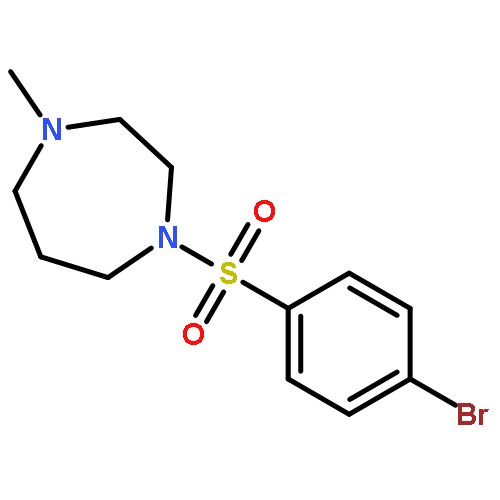
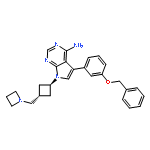
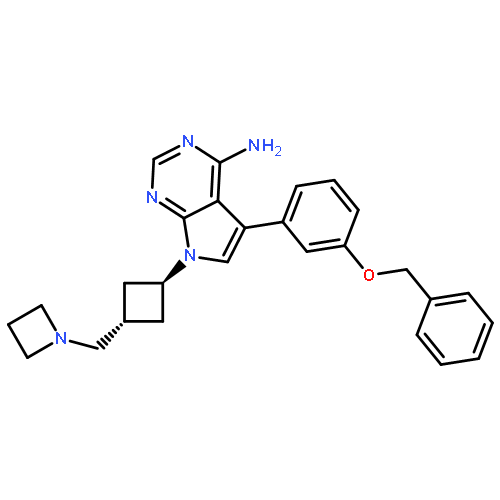
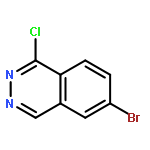




![N-[(3Z)-2-OXO-3-[PHENYL-[4-(PIPERIDIN-1-YLMETHYL)ANILINO]METHYLIDENE]-1H-INDOL-5-YL]ETHANESULFONAMIDE](http://img.cochemist.com/ccimg/422600/422513-13-1.png)
![N-[(3Z)-2-OXO-3-[PHENYL-[4-(PIPERIDIN-1-YLMETHYL)ANILINO]METHYLIDENE]-1H-INDOL-5-YL]ETHANESULFONAMIDE](http://img.cochemist.com/ccimg/422600/422513-13-1_b.png)




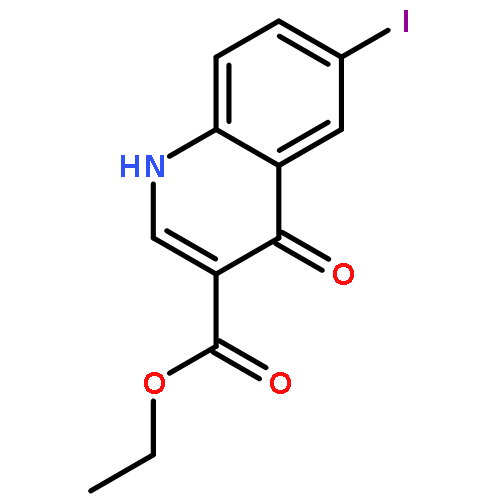




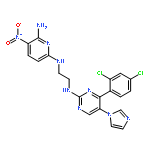
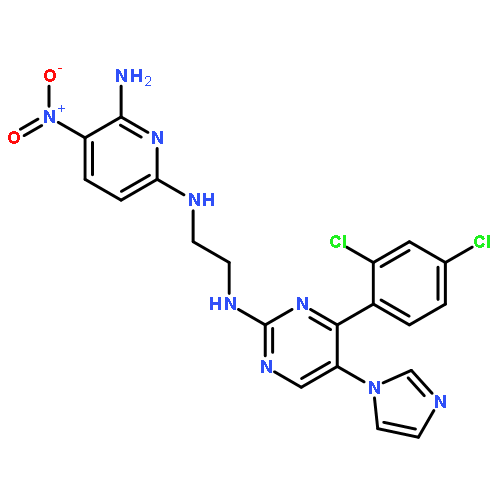


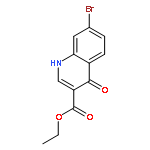
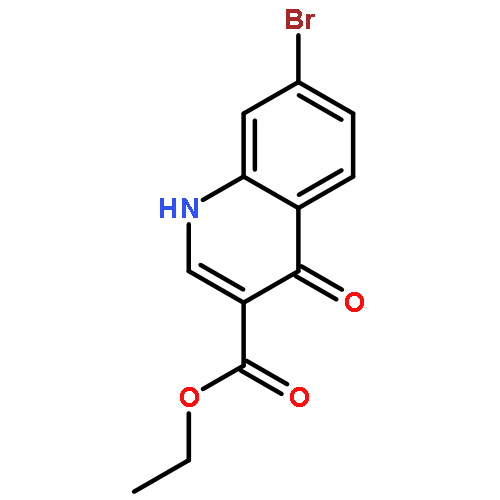


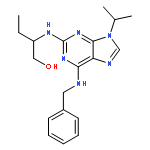
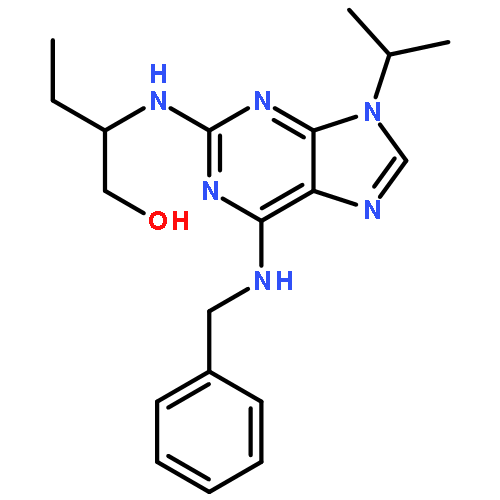




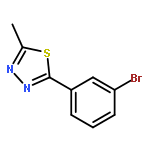
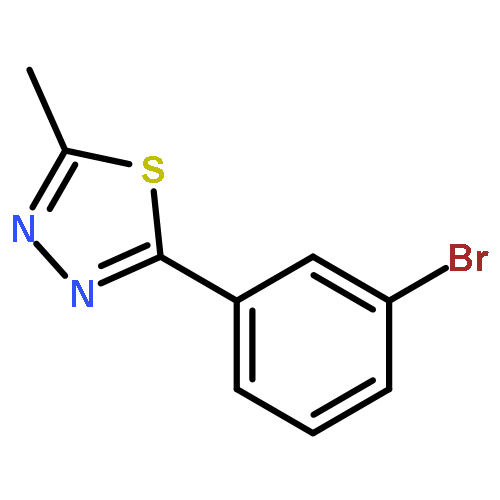






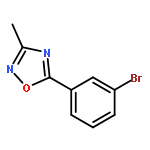
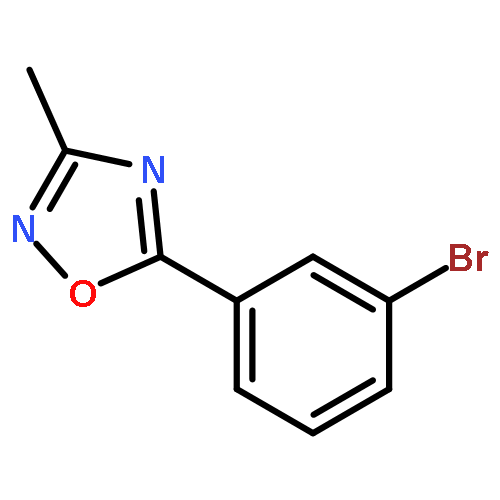
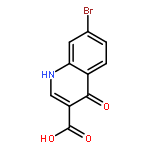
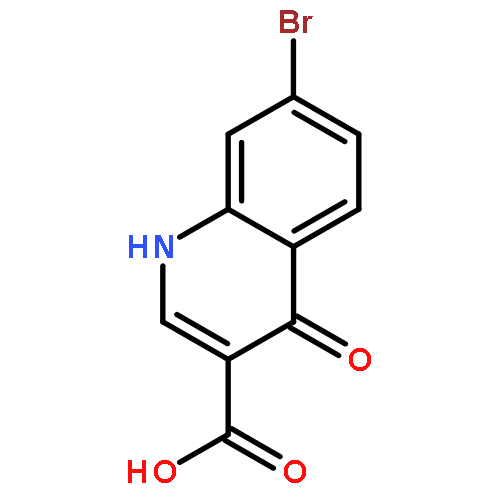
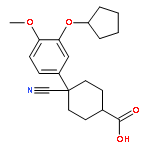
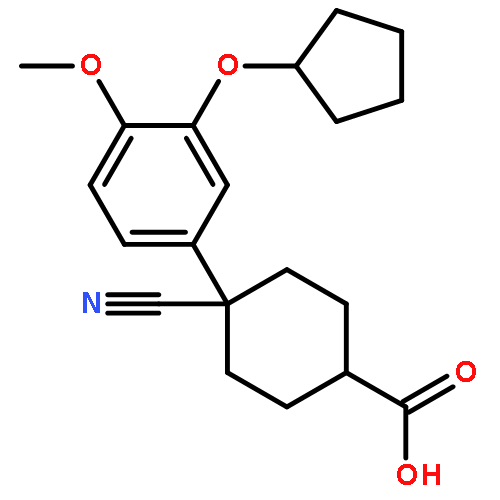


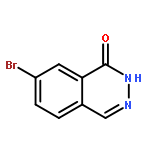















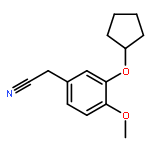
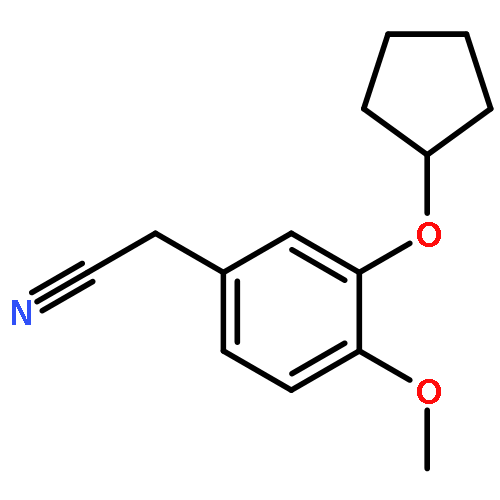
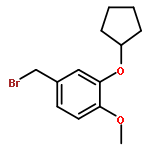
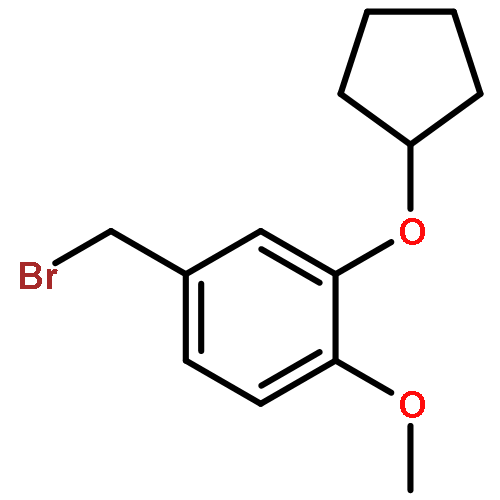




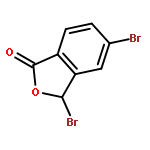
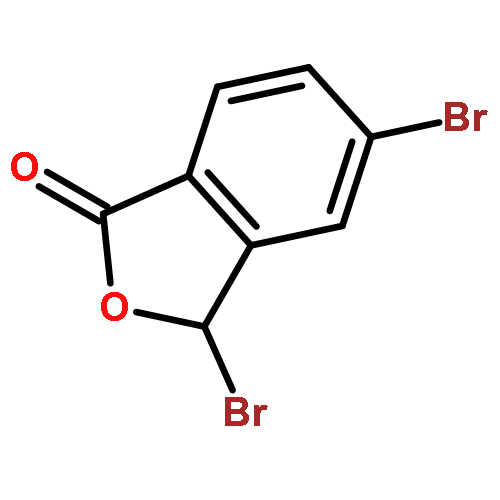


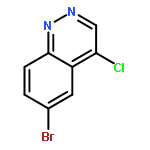
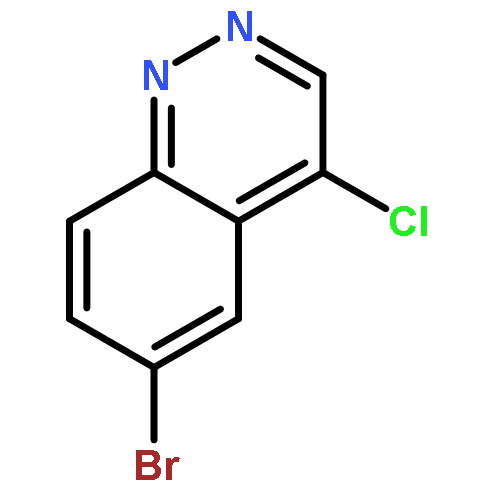
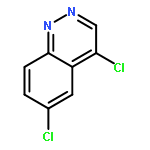
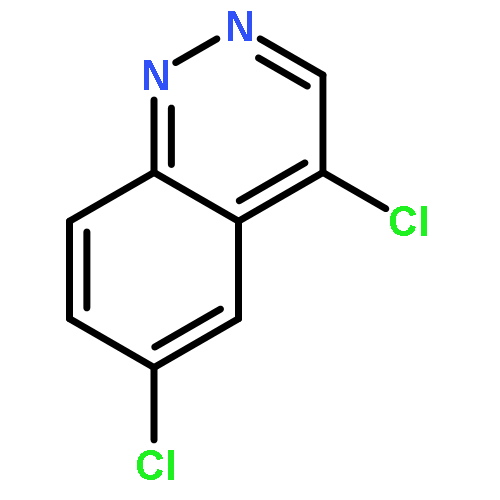


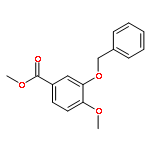
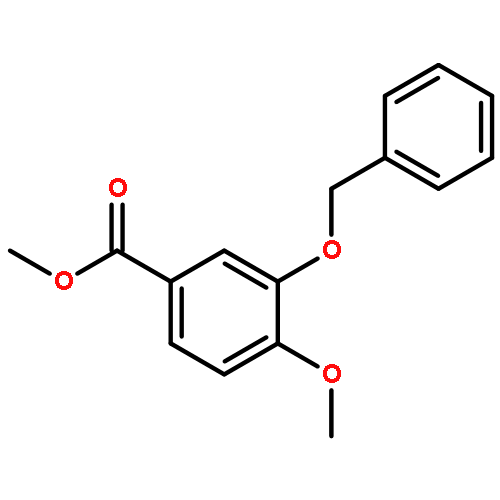



![PROPANEDIOIC ACID, [[(4-IODOPHENYL)AMINO]METHYLENE]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/40200/40107-05-9.png)
![PROPANEDIOIC ACID, [[(4-IODOPHENYL)AMINO]METHYLENE]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/40200/40107-05-9_b.png)



![Thieno[3,2-d]pyrimidine](http://img.cochemist.com/ccimg/300/272-68-4.png)
![Thieno[3,2-d]pyrimidine](http://img.cochemist.com/ccimg/300/272-68-4_b.png)


![6-Bromo-4-chlorothieno[3,2-d]pyrimidine](http://img.cochemist.com/ccimg/225400/225385-03-5.png)
![6-Bromo-4-chlorothieno[3,2-d]pyrimidine](http://img.cochemist.com/ccimg/225400/225385-03-5_b.png)



