Co-reporter:Houzong Yao, Fengyou Liu, Jinglei Chen, Yan Li, Jinhao Cui, Chunhua Qiao
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 5) pp:1386-1390
Publication Date(Web):1 March 2016
DOI:10.1016/j.bmcl.2016.01.075
Although the antischistosomal activities of N,N′-arylurea analogs were reported, systematic structure–activity relationships have not been conducted. In this Letter, we reported the design, synthesis and evaluation of 45 N,N′-arylurea analogs. Among these prepared compounds, 13 compounds were urea linker modified and 32 were N,N′-arylurea derivatives. The activity evaluation revealed 12 analogs exhibited IC50 values lower than 22.6 μM, and 7 of them had IC50 less than 10 μM against the juvenile Schistosoma japonicum in vitro. Their worm killing potency was even higher against adult worm. Unfortunately, low to moderate worm burden reduction of 0–33.4% was recorded after administration of a single oral dose of 200 mg/kg or 400 mg/kg to mice harboring S. japonicum.The IC50s of seven potent N,N′-arylurea compounds against juvenile and adult S. japonicum. Compounds 16 and 38 showed higher activities than the positive control MMV665852.
Co-reporter:Peng Ji, Xin Xu, Shuhua Ma, Junchao Fan, Qiang Zhou, Xinliang Mao, and Chunhua Qiao
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 9) pp:1010
Publication Date(Web):July 27, 2015
DOI:10.1021/acsmedchemlett.5b00228
Signal transducer and activator of transcription 3 (STAT3) is considered to be an attractive therapeutic target for cancer therapy. In this study, a series of 2-carbonylbenzo[b]thiophene 1,1-dioxide derivatives (CBT) were designed to inhibit the STAT3 SH2 domain phosphorylation site Try 705. We demonstrated that incorporation of basic flexible groups through amide bond linkage to benzo[b]thiophene 1,1-dioxide (BTP) achieved compounds with higher antiproliferative potency than BTP itself. The most potent compound 6o, as indicated from luciferase reporter gene assay, inhibited the STAT3 pathway by decreasing the phosphorylation level of STAT3 Tyr705, while the phosphorylation level of other upstream tyrosine kinases in this pathway was not significantly inhibited. Compound 6o was also shown to trigger ROS generation and accumulation, thus consequently attributed partially to the observed cell apoptosis. This study provided important structural information for the development of inhibitors targeting the STAT3 pathway.Keywords: 2-carbonylbenzo[b]thiophene 1,1-dioxide derivatives; antiproliferative activity; apoptosis; inhibitor; reactive oxygen species; STAT3;
Co-reporter:Jing Yang, Yanli Liu, Chengwen Xue, Wei Yu, Jian Zhang, Chunhua Qiao
European Journal of Medicinal Chemistry 2014 Volume 86() pp:235-241
Publication Date(Web):30 October 2014
DOI:10.1016/j.ejmech.2014.08.061
•Sixteen Glaucocalyxin (GLA) derivatives were prepared through Michael addition reaction between GLA and amines.•MTT assay revealed that the resulting GLA derivatives retained antiproliferative effects against six cancer cell lines.•These GLA derivatives were shown to exhibit reduced cytotoxicity against normal cell.•The plasma incubation analysis confirmed the prodrug property of these GLA derivatives.•These prodrugs exhibited markedly enhanced plasma stability.A series of Mannich base type derivatives of Glaucocalyxin A (GLA) were designed and prepared. The cytotoxicity of these compounds was evaluated against six tumor cell lines (SMMC-7721, B16, SGC-7901, A549, KB, HL-60). Most compounds exhibited potent antiproliferative effects with low micromolar IC50 values. Compound 1 with para methyl benzyl amine moiety and compound 16 with cyclohexylamine moiety displayed the highest inhibition efficacy. Significantly, the cytotoxicity of compound 1 was much lower than GLA against the normal human liver cell (HL-7702). The in vitro stability assay revealed that transformation of GLA to Mannich base type derivatives improved the compound stability in rat plasma. Finally, decomposition product analysis supported that compound 1 could act as a prodrug and release GLA in the intracellular environment.
Co-reporter:Lili Wang, Ying-Ying Zhang, Lei Wang, Feng-you Liu, Ling-Ling Cao, Jing Yang, Chunhua Qiao, Yonghao Ye
European Journal of Medicinal Chemistry 2014 80() pp: 535-542
Publication Date(Web):
DOI:10.1016/j.ejmech.2014.04.058
Co-reporter:Zhixiang Xu, Wei Yin, Leonardo K. Martinelli, Joanna Evans, Jinglei Chen, Yang Yu, Daniel J. Wilson, Valerie Mizrahi, Chunhua Qiao, Courtney C. Aldrich
Bioorganic & Medicinal Chemistry 2014 22(5) pp: 1726-1735
Publication Date(Web):
DOI:10.1016/j.bmc.2014.01.017
Co-reporter:Lili Wang, Cong Li, Yingying Zhang, Chunhua Qiao, and Yonghao, Ye
Journal of Agricultural and Food Chemistry 2013 Volume 61(Issue 36) pp:8632-8640
Publication Date(Web):August 12, 2013
DOI:10.1021/jf402388x
Forty-four benzofuroxan derivatives were designed and prepared as antifungal agents. Their structures were characterized by 1H NMR, 13C NMR, and HRMS. Their antifungal activities were tested in vitro with four important phytopathogenic fungi, namely, Rhizoctonia solani, Sclerotinia sclerotiorum, Fusarium graminearum and Phytophthora capsici, using the mycelium growth inhibition method. Compound A5 displayed the maximum antifungal activity against F. graminearum (IC50 = 1.1 μg/mL, which is about 2-fold higher than that of the well-known positive control carbendazim (IC50 = 0.5 μg/mL). A14 exhibited high antifungal effect against both S. sclerotiorum and F. graminearum Sehw., with IC50 values of 2.52 and 3.42 μg/mL, respectively. Among 14 benzofuroxan derivatives with substitutions at the R2 and R3 positions of the phenyl ring (B series), 7 compounds displayed strong growth inhibition against R. solani (IC50 ≤ 3.0 μg/mL). Analysis of the structure–activity relationship data of these compounds revealed that (1) introduction of an electron-donating amino group to the R2 position of the phenyl ring favors antifungal activity against R. solani and (2) the presence of a nitro group at the R4 position and substituent variation at the R1 position of the phenyl ring can result in good antifungal candidates against F. graminearum Sehw. Overall, the benzofuroxan was discovered as a novel scaffold for the development of fungicides. Significantly, A14 was demonstrated to successfully suppress disease development in S. sclerotiorum infected cole in vivo.
Co-reporter:Zhi-xia Wang;Jing-lei Chen
Chemical Biology & Drug Design 2013 Volume 82( Issue 2) pp:216-225
Publication Date(Web):
DOI:10.1111/cbdd.12153
A series of aromatic ring-modified praziquantel derivatives were prepared and evaluated against juvenile and adult stage of Schistosoma japonicumin. Several analogs comparable in activity to the drug praziquantel have been identified based on in vitro and in vivo japonuicum schistosomes worm viability assay. Structure and activity relationship of these praziquantel aromatic ring-modified compounds was revealed. Specifically, a compound in which a bromine has been introduced in the aromatic ring of praziquantel demonstrated close antischistosomal activity to praziquantel in vivo.
Co-reporter:Jinglei Chen, Weizhen Sun, Jingjing Yang, Huan Sun, Zhixia Wang, Lanlan Dong, Chunhua Qiao, Chao-ming Xia
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 13) pp:3785-3787
Publication Date(Web):1 July 2013
DOI:10.1016/j.bmcl.2013.04.085
Analogues of pyrrolo-[1,2,5]benzothiadiazepine were prepared and evaluated against Schistosoma japonica. The biological data revealed that most benzothiazepine derivatives show anti-schistosomal activity to some extent, while α-chloronation of the title compound and another bioisosteric derivative pyrrolo-[1,2,5]benzodiazepine displayed the most distinct worm killing activity. This study proved that benzodiazepine may serve as a novel structural skeleton for the development of anti-schistosomal agents.
Co-reporter:Wei Yin
Journal of Heterocyclic Chemistry 2013 Volume 50( Issue 6) pp:1290-1293
Publication Date(Web):
DOI:10.1002/jhet.1560
Epigoitrin is one of the major components of several natural species, including Isatis indigotica Fort, turnip, and cabbage. It presents antithyroid and antivirus activities. Here, we report an efficient and practical method for the chemical synthesis of epigoitrin from commercially available (R)-(+)-4-hydroxy-γ-butyrolactone.
Co-reporter:Huan-qiu Li, Jing Yang, Shuhua Ma, Chunhua Qiao
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 14) pp:4194-4200
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmc.2012.05.079
A series of novel rhodanine-based acylsulfonamide derivatives were designed, synthesized, and evaluated as small-molecule inhibitors of anti-apoptotic Bcl-2 protein. These compounds exhibit potent antiproliferative activity in three human tumor cell lines (Hep G2, PC-3 and B16-F10). Among them, the most potent compounds 10 and 11 bind to Bcl-2 with a Ki of 20 and 25 nM, respectively. Docking studies demonstrated that these two compounds orient similarly at the binding site of Bcl-2, and the calculated binding affinities (Glide XP score) of compound 10 is more negative than that of compound 11. The binding interactions of compounds with high binding affinity to Bcl-2 protein were analyzed.Rhodanine-based sulfonamides were developed as antagonists of anti-apoptotic Bcl-2 protein.
Co-reporter:Wen-wen Duan, Si-jie Qiu, Yue Zhao, Huan Sun, Chunhua Qiao, Chao-ming Xia
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 4) pp:1587-1590
Publication Date(Web):15 February 2012
DOI:10.1016/j.bmcl.2011.12.133
A praziquantel analog 10-hydroxy praziquantel and eight praziquantel/peroxide conjugates were synthesized. The biological activity of these compounds was evaluated against juvenile and adult stages of Schistosoma japonicum. Unlike praziquantel, 10-hydroxy praziquantel exhibits activity against both juvenile and adult Schistosoma japonicumin. All hybrid compounds displayed modest to significant worm killing activity. The present study has important significance for the development of hybrid antischistosomal drugs.Praziquantel derivatives with activity against both juvenile and adult Schistosoma j. in vitro.
Co-reporter:Jian-Zhong Wu, Zhixia Wang, Chunhua Qiao
Tetrahedron Letters 2012 Volume 53(Issue 9) pp:1153-1155
Publication Date(Web):29 February 2012
DOI:10.1016/j.tetlet.2011.12.102
Total synthesis of stagonolide C using chiral pool strategy is described. The two key intermediates were prepared from l-glutamic acid and d-glucono-1,5-lactone, followed by Julia–Lythgoe olefination and Yamaguchi esterification to afford the target compound in an efficient way.
Co-reporter:Jing Yang, Zhengchao Tu, Xin Xu, Jinfeng Luo, Xing Yan, Chongzhao Ran, Xinliang Mao, Ke Ding, Chunhua Qiao
Bioorganic & Medicinal Chemistry Letters (15 March 2017) Volume 27(Issue 6) pp:
Publication Date(Web):15 March 2017
DOI:10.1016/j.bmcl.2017.02.023
•4-Anilinoquinazoline and endoperoxide conjugates exhibited high sensitivity to gefitinib resistant cancer cells.•Some conjugates retained EGFR inhibitory activity.•Selectivity toward cancer cell vs normal cell was observed for some conjugates.In the present study, endoperoxide and 4-anilinoqnazoline were conjugated to obtain a series of compounds. These conjugates exhibited high antiproliferative potency against a number of cancer cell lines, including the epidermal growth factor receptor (EGFR) L858R/T790M mutant cell. Compound 5 was selected as a representative for mechanistic study. Further experiments revealed the conjugate’s reactive oxygen species (ROS) generating ability, apoptosis inducing activity and involvement in EGFR downstream signaling pathways.Gefinitib resistant cancer cells exhibited high sensitivity to conjugates of 4-anilinoquinazoline with endoperoxide.

![N-[3,5-Bis(trifluoromethyl)phenyl]-N′-[(8a,9S)-10,11-dihydro-6′-methoxy-9-cinchonanyl]thiourea](/data/chemimg/1588800/852913-19-0.png)
![N-[3,5-Bis(trifluoromethyl)phenyl]-N′-[(8a,9S)-10,11-dihydro-6′-methoxy-9-cinchonanyl]thiourea](/data/chemimg/1588800/852913-19-0_b.png)






![2-(1,4-Dioxaspiro[4.5]decan-8-yl)ethanol](http://img.cochemist.com/ccimg/135800/135761-76-1.png)
![2-(1,4-Dioxaspiro[4.5]decan-8-yl)ethanol](http://img.cochemist.com/ccimg/135800/135761-76-1_b.png)
![7,8,15,16-Tetraoxadispiro[5.2.5.2]hexadecane-3-acetic acid](http://img.cochemist.com/ccimg/923300/923267-21-4.png)
![7,8,15,16-Tetraoxadispiro[5.2.5.2]hexadecane-3-acetic acid](http://img.cochemist.com/ccimg/923300/923267-21-4_b.png)
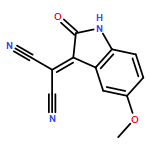
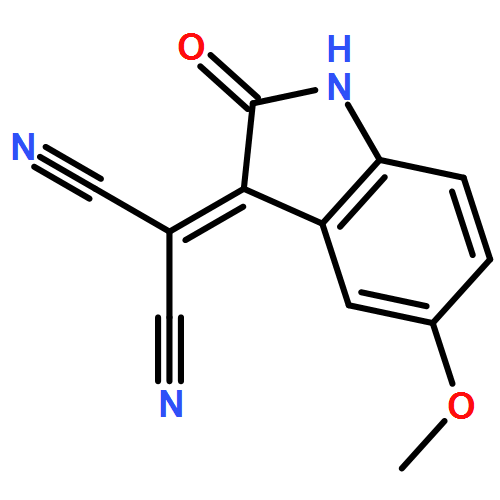
![Piperidine, 1-[(5-aminobenzo[b]thien-2-yl)carbonyl]-](http://img.cochemist.com/ccimg/832200/832102-94-0.png)
![Piperidine, 1-[(5-aminobenzo[b]thien-2-yl)carbonyl]-](http://img.cochemist.com/ccimg/832200/832102-94-0_b.png)








![1-[4-[(4-NITRO-2,1,3-BENZOXADIAZOL-7-YL)AMINO]PHENYL]ETHANONE](http://img.cochemist.com/ccimg/314000/313966-78-8.png)
![1-[4-[(4-NITRO-2,1,3-BENZOXADIAZOL-7-YL)AMINO]PHENYL]ETHANONE](http://img.cochemist.com/ccimg/314000/313966-78-8_b.png)
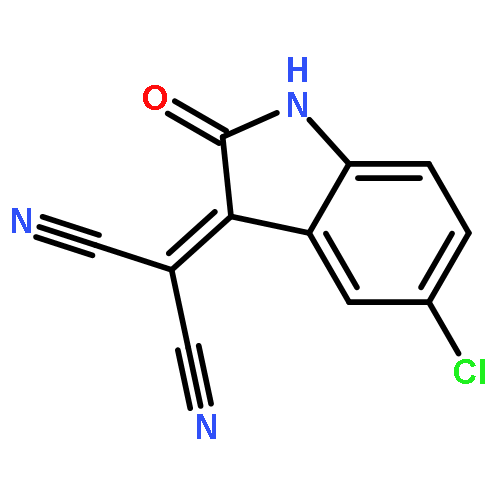
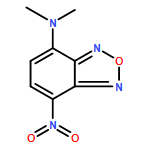
![4-Nitro-7-(piperazin-1-yl)benzo[c][1,2,5]oxadiazole](/data/chemimg/461700/139332-66-4.png)
![4-Nitro-7-(piperazin-1-yl)benzo[c][1,2,5]oxadiazole](/data/chemimg/461700/139332-66-4_b.png)
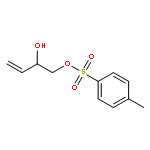

![2-(1,4-DIOXASPIRO[4.5]DECAN-8-YL)ACETIC ACID](http://img.cochemist.com/ccimg/134200/134136-04-2.png)
![2-(1,4-DIOXASPIRO[4.5]DECAN-8-YL)ACETIC ACID](http://img.cochemist.com/ccimg/134200/134136-04-2_b.png)
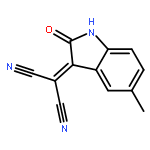
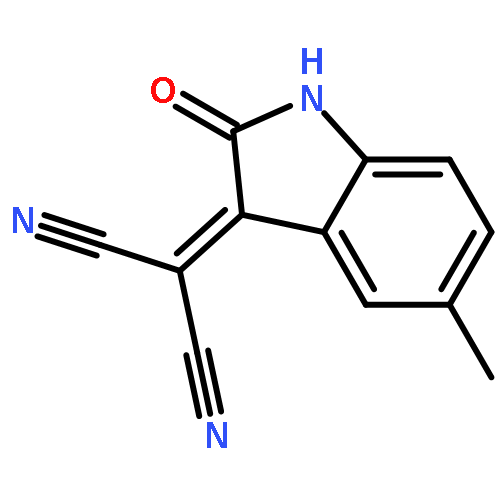










![Piperidine, 1-[(5-nitrobenzo[b]thien-2-yl)carbonyl]-](http://img.cochemist.com/ccimg/97600/97528-67-1.png)
![Piperidine, 1-[(5-nitrobenzo[b]thien-2-yl)carbonyl]-](http://img.cochemist.com/ccimg/97600/97528-67-1_b.png)










![Urea, N-[(4-chlorophenyl)methyl]-N'-(3,4-dichlorophenyl)-](http://img.cochemist.com/ccimg/65700/65608-80-2.png)
![Urea, N-[(4-chlorophenyl)methyl]-N'-(3,4-dichlorophenyl)-](http://img.cochemist.com/ccimg/65700/65608-80-2_b.png)


































![2,1,3-Benzoxadiazole,4-nitro-7-[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/16400/16322-24-0.png)
![2,1,3-Benzoxadiazole,4-nitro-7-[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/16400/16322-24-0_b.png)
![4-Nitrobenzo[c][1,2,5]oxadiazole](http://img.cochemist.com/ccimg/16400/16322-19-3.png)
![4-Nitrobenzo[c][1,2,5]oxadiazole](http://img.cochemist.com/ccimg/16400/16322-19-3_b.png)













![METHYL 4-[2-HYDROXY-3-(ISOPROPYLAMINO)PROPOXY]BENZOATE HYDROCHLOR<WBR />IDE (1:1)](http://img.cochemist.com/ccimg/7100/7019-01-4.png)
![METHYL 4-[2-HYDROXY-3-(ISOPROPYLAMINO)PROPOXY]BENZOATE HYDROCHLOR<WBR />IDE (1:1)](http://img.cochemist.com/ccimg/7100/7019-01-4_b.png)
![5-Nitrobenzo[b]thiophene-2-carboxylic acid](http://img.cochemist.com/ccimg/6400/6345-55-7.png)
![5-Nitrobenzo[b]thiophene-2-carboxylic acid](http://img.cochemist.com/ccimg/6400/6345-55-7_b.png)