Co-reporter:Li Li, Deshuai Yang, Trevor R. Fisher, Qi Qiao, Zhen Yang, Na Hu, Xiangshu Chen, and Liangliang Huang
Langmuir October 24, 2017 Volume 33(Issue 42) pp:11543-11543
Publication Date(Web):July 21, 2017
DOI:10.1021/acs.langmuir.7b01537
The loading-dependent diffusion behavior of CH4, CO2, SO2, and their binary mixtures in ZIF-10 has been investigated in detail by using classical molecular dynamics simulations. Our simulation results demonstrate that the self-diffusion coefficient Di of CH4 molecules decreases sharply and monotonically with the loading while those of both CO2 and SO2 molecules initially display a slight increase at low uptakes and follow a slow decrease at high uptakes. Accordingly, the interaction energies between CH4 molecules and ZIF-10 remain nearly constant regardless of the loading due to the absence of hydrogen bonds (HBs), while the interaction energies between CO2 (or SO2) and ZIF-10 decease rapidly with the loading, especially at small amounts of gas molecules. Such different loading-dependent diffusion and interaction mechanisms can be attributed to the relevant HB behavior between gas molecules and ZIF-10. At low loadings, both the number and strength of HBs between CO2 (or SO2) molecules and ZIF-10 decrease obviously as the loading increases, which is responsible for the slight increase of their diffusion coefficients. However, at high loadings, their HB strength increases with the loading. Similar loading-dependent phenomena of diffusion, interaction, and HB behavior can be observed for CH4, CO2, and SO2 binary mixtures in ZIF-10, only associated with some HB competition between CO2 and SO2 molecules in the case of the CO2/SO2 mixture.
Co-reporter:Li Li, Deshuai Yang, Trevor R. Fisher, Qi Qiao, Zhen Yang, Na Hu, Xiangshu Chen, and Liangliang Huang
Langmuir October 24, 2017 Volume 33(Issue 42) pp:11543-11543
Publication Date(Web):July 21, 2017
DOI:10.1021/acs.langmuir.7b01537
The loading-dependent diffusion behavior of CH4, CO2, SO2, and their binary mixtures in ZIF-10 has been investigated in detail by using classical molecular dynamics simulations. Our simulation results demonstrate that the self-diffusion coefficient Di of CH4 molecules decreases sharply and monotonically with the loading while those of both CO2 and SO2 molecules initially display a slight increase at low uptakes and follow a slow decrease at high uptakes. Accordingly, the interaction energies between CH4 molecules and ZIF-10 remain nearly constant regardless of the loading due to the absence of hydrogen bonds (HBs), while the interaction energies between CO2 (or SO2) and ZIF-10 decease rapidly with the loading, especially at small amounts of gas molecules. Such different loading-dependent diffusion and interaction mechanisms can be attributed to the relevant HB behavior between gas molecules and ZIF-10. At low loadings, both the number and strength of HBs between CO2 (or SO2) molecules and ZIF-10 decrease obviously as the loading increases, which is responsible for the slight increase of their diffusion coefficients. However, at high loadings, their HB strength increases with the loading. Similar loading-dependent phenomena of diffusion, interaction, and HB behavior can be observed for CH4, CO2, and SO2 binary mixtures in ZIF-10, only associated with some HB competition between CO2 and SO2 molecules in the case of the CO2/SO2 mixture.
Co-reporter:Yiping Huang, Zheng Wan, Zhen Yang, Yuanhui Ji, Li Li, Deshuai Yang, Meihua Zhu, and Xiangshu Chen
Journal of Chemical & Engineering Data August 10, 2017 Volume 62(Issue 8) pp:2340-2340
Publication Date(Web):July 20, 2017
DOI:10.1021/acs.jced.7b00205
The detailed hydrogen bond (HB) behavior of ethylammonium nitrate (EAN) ionic liquid (IL)–water mixtures with different water concentrations has been investigated at a molecular level by using classical molecular dynamics simulations. The simulation results demonstrate that the increasing water concentration can weaken considerably all cation–anion, cation–water, anion–water, and water–water HBs in EAN–water mixtures, and the corresponding HB networks around cations, anions, and water molecules also change significantly with the addition of water. Meanwhile, both the translational and the rotational motions of anions, cations, and water molecules are found to be much faster as the water concentration increases. On the other hand, the order of their HB strength is found to be cation–anion > anion–water > cation–water > water–water at low water mole fractions (<38%), while the corresponding order is cation–anion > cation–water > anion–water > water–water at high water mole fractions (>38%). The opposite orders of anion–water and cation–water HBs at low and high water concentrations, as well as the different changes of HB networks around cations and anions, should be responsible for the increasing deviation in diffusion coefficient between cations and anions with the water concentration, which is favorable to the cation–anion dissociation. In addition, the competing effect between ionic mobility and ionic concentration leads to that the ionic conductivity of EAN–water mixtures initially increases with the water mole fraction and follows a sharp decrease beyond 90%. Our simulation results provide a molecular-level concentration-dependent HB networks and dynamics, as well as their relationship with unique structures and dynamics in protic IL–water mixtures.
Co-reporter:Fangjia Fu, Yunzhi Li, Zhen YangGuobing Zhou, Yiping Huang, Zheng Wan, Xiangshu Chen, Na Hu, Wei Li, Liangliang Huang
The Journal of Physical Chemistry C 2017 Volume 121(Issue 1) pp:
Publication Date(Web):December 21, 2016
DOI:10.1021/acs.jpcc.6b11043
Here we report a series of classical molecular dynamics simulations for the icosahedral Au nanoparticles with four different diameters of 1.0, 1.4, 1.8, and 2.3 nm in 1-butyl-3-methylimidazolium tetrafluoroborate ([bmim][BF4]) room-temperature ionic liquid (RTIL). Our simulation results reveal for the first time a size-dependent stabilization mechanism of the Au nanoparticles in the [bmim][BF4] RTIL, which may help to clarify the relevant debate on the stabilization mechanism from various experimental observations. By comparison, the alkyl chains in the [bmim]+ cations are found to dominate the stabilization of the smallest Au13 nanoparticle in the RTIL while the imidazolium rings should be mainly responsible for the stabilization of other larger nanoparticles in the RTIL. Compared to the [bmim]+ cations, the [BF4]− anions are found to have an indirect influence on stabilizing the Au nanoparticles in the RTIL because of the weak interaction between the Au nanoparticles and the anions. However, such differences in the stabilization mechanism between the small and the large Au nanoparticles can be attributed to the unique hydrogen bond (HB) network between the cations and the anions in the first solvation shell. Meanwhile, increasing the particle size can lead to the enhanced HBs on the surface of Au nanoparticles, so slower rotational motions and more pronounced orientation distribution of cations can be observed around the larger nanoparticles. Our simulation results in this work provide a molecular-level understanding of the unique size-dependent stabilization mechanism of the Au nanoparticles in the imidazolium-based RTILs.
Co-reporter:Yiping Huang, Guobing Zhou, Yunzhi Li, Zhen Yang, Man Shi, Xueping Wang, Xiangshu Chen, Fei Zhang, Wei Li
Chemical Physics 2016 Volume 472() pp:105-111
Publication Date(Web):15 June 2016
DOI:10.1016/j.chemphys.2016.03.020
Highlights
- •
Hydrogen bonds play an essential role in determining dynamics properties of protic ionic liquids.
- •
Temperature has an important influence on dynamics properties rather than structures.
- •
Both anions and cations are found to display an obvious sub-diffusive behavior regardless of the temperature.
Co-reporter:Guobing Zhou
The Journal of Physical Chemistry C 2016 Volume 120(Issue 9) pp:5033-5041
Publication Date(Web):February 22, 2016
DOI:10.1021/acs.jpcc.6b00307
The structures and relevant vibrational spectra of an ethylammonium nitrate (EAN) ionic liquid (IL) confined in single-walled carbon nanotubes (SWCNTs) with various diameters have been investigated in detail by using classical molecular dynamics simulation. Our simulation results demonstrate that the EAN IL confined in larger SWCNTs can form well-defined multishell structures with an additional cation chain located at the center. However, a different single-shell hollow structure has been found for both the cations and the anions in the 1 nm SWCNT. For the cations confined in SWCNTs, the CH3 groups stay closer to the nanotube walls because of their solvophobic nature, while the NH3+ groups prefer to point toward the central axis. Accordingly, the NO3– anions tend to lean on the SWCNT surface with three O atoms facing the central axis to form hydrogen bonds (HBs) with the NH3+ groups. In addition, in the 1 nm SWCNT, the CH3 groups of cations exhibit an obvious blue shift of around 16 cm–1 for the C–H stretching mode with respect to the bulk value, and the N–H stretching mode of NH3+ groups is split into two characteristic peaks with one peak appearing at a higher frequency. Such a blue shift is attributed to the existence of more free space for the C–H bonds of confined CH3 groups, while the splitting phenomenon is due to the fact that more than 60% of the confined NH3+ groups have one dangling N–H bond. For the anions confined in the 1 nm SWCNT, the N–O stretching mode of NO3– has a maximum red shift of around 24 cm–1 with respect to the bulk value, which is attributed to enhanced HBs between anions and cations. Our simulation results reveal a molecular-level correlation between confined structural configurations and the corresponding vibrational spectra changes for the ILs confined in nanometer scale environments.
Co-reporter:Guobing Zhou, Zhen Yang, Fangjia Fu, Yiping Huang, Xiangshu Chen, Zhanghui Lu, and Na Hu
Industrial & Engineering Chemistry Research 2015 Volume 54(Issue 33) pp:8166-8174
Publication Date(Web):August 4, 2015
DOI:10.1021/acs.iecr.5b01624
Molecular dynamics simulations have been performed to explore the solvation structures and vibrational spectra of an ethylammonium nitrate (EAN) ionic liquid (IL) around various single-walled carbon nanotubes (SWNTs). Our simulation results demonstrate that both cations and anions show a cylindrical double-shell solvation structure around the SWNTs regardless of the nanotube diameter. In the first solvation shell, the CH3 groups of cations are found to be closer to the SWNT surface than the NH3+ groups because of the solvophobic nature of the CH3 groups, while the NO3– anions tend to lean on the nanotube surface, with three O atoms facing the bulk EAN. On the other hand, the intensities of both C–H (the CH3 group of the cation) and N–O (anion) asymmetric stretching bands at the EAN/SWNT interface are found to be slightly higher than the corresponding bulk values owing to the accumulation and orientation of cations and anions in the first solvation shell. More interestingly, the N–O stretching band exhibits a red shift of around 10 cm–1 with respect to the bulk value, which is quite contrary to the blue shift of the O–H stretching band of water molecules at the hydrophobic interfaces. Such a red shift of the N–O stretching mode can be attributed to the enhanced hydrogen bonds (HBs) of the NO3– anions in the first solvation shell. Our simulation results provide a molecular-level understanding of the interfacial vibrational spectra of an EAN IL on the SWNT surface and their connection with the relevant solvation structures and interfacial HBs.
Co-reporter:Zhen Yang
The Journal of Physical Chemistry C 2015 Volume 119(Issue 4) pp:1768-1781
Publication Date(Web):January 13, 2015
DOI:10.1021/jp506875m
Molecular dynamics simulations have been performed to systematically investigate the structure and dynamics properties, hydrogen bond (HB) dynamics, and far-infrared (far-IR) spectra of hydration water molecules around the mixed monolayer-protected Au nanoparticles (MPANs) with different ligand compositions and length. Our simulation results demonstrate that the translational and rotational motions of hydration water molecules in the proximity of charged terminal NH3+ and COO– groups are suppressed significantly with respect to the bulk water. Compared to the bulk water, meanwhile, longer structural relaxation times of hydration H2O–H2O HBs indicate enhanced strength of H2O–H2O HBs at the interface of mixed MPANs. Accordingly, these hydration water molecules around the charged terminal groups can exhibit a considerable blue-shift in far-IR spectra for all ligand compositions and length studied here. A series of detailed HB analyses manifest that above restricted behavior of hydration water molecules can be attributed to the stronger H2O–NH3+ and H2O–COO– HBs and the corresponding structural relaxation times are much greater than those of hydration H2O–H2O HBs. Furthermore, we find that increasing ligand length can affect much the morphology of self-assemble monolayers in water owing to enhanced hydrophobic interactions between alkane chains and water molecules and favor the translational mobility of hydration water molecules owing to weaken electrostatic interactions. Unlike the translational motions, our comparison results among different ligand lengths clearly confirm that the rotational relaxation of hydration water molecules should be dominated by the local and directional HBs at the interfaces, rather than the previous explanation of the ratio between hydrophobic/hydrophilic exposed regions. More importantly, our simulations reveal at a molecular level that the ligand composition has a little influence on the structure, dynamics, HBs, and far-IR spectra of hydration water molecules around the mixed MPANs mainly due to the comparable strength between H2O–NH3+ and H2O–COO– HBs.
Co-reporter:Zhen Yang, Hao Lin, Tian Gui, Rong-Fei Zhou, Xiang-Shu Chen
Chinese Chemical Letters 2014 Volume 25(Issue 1) pp:107-110
Publication Date(Web):January 2014
DOI:10.1016/j.cclet.2013.09.009
The infrared (IR) spectra of the N-methylacetamide molecule in water are calculated by using the MD simulation with high-level QM/MM corrections. The B3LYP and MP2 levels with 6-311++G** basis set are used for the QM region, respectively. Our results show all IR spectra at the B3LYP level are well consistent with the corresponding MP2 results. A dynamical charge fluctuation is observed for each atom along the simulation trajectories due to the electrostatic polarization (EP) effects from surrounding solvent environment. We find that the QM/MM corrected IR spectra satisfactorily reproduce the experimental vibrational features of amide I–III modes.Electrostatic polarization has a significant influence on the IR spectroscopy of peptides.

![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)













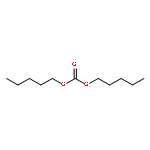
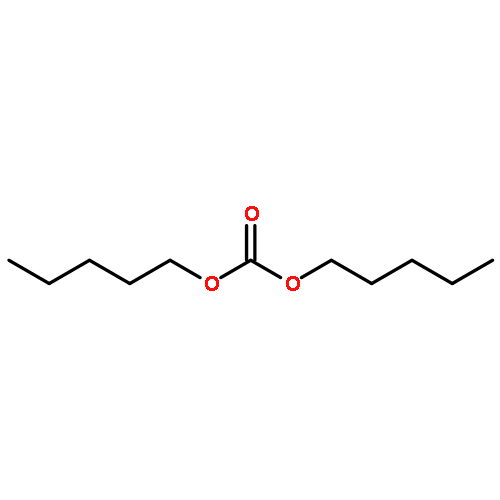
![1,5-Dioxaspiro[5.5]undecane,2-methyl-](http://img.cochemist.com/ccimg/6500/6413-26-9.png)
![1,5-Dioxaspiro[5.5]undecane,2-methyl-](http://img.cochemist.com/ccimg/6500/6413-26-9_b.png)

![1,4-Dioxaspiro[4.5]decane,2-methyl-](http://img.cochemist.com/ccimg/4800/4722-68-3.png)
![1,4-Dioxaspiro[4.5]decane,2-methyl-](http://img.cochemist.com/ccimg/4800/4722-68-3_b.png)