Co-reporter:Xue-Peng Wang, Xian-Bin Li, Nian-Ke Chen, Qi-Dai Chen, ... Hong-Bo Sun
Acta Materialia 2017 Volume 136(Volume 136) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.actamat.2017.07.006
GeSbTe alloys have the ability of rapidly transforming between amorphous and crystalline phases. Therefore, they can be used in the non-volatile phase change memory. Recently, a vacancy-ordered cubic Ge2Sb2Te5 (VOC GST) phase change material where the vacancies are highly ordered in the (111) plane, has been experimentally demonstrated by STEM. However, studies are mainly on the structural characterization, rather than on the phase change behavior and possible applications of the VOC GST. Here, using first-principles molecular dynamic simulations, we study the melt-quenched amorphization process and its possible applications. We find that the VOC GST exhibits a quasi-two-dimensional amorphization process that is triggered by the diffusion of Ge atoms but not others. A partial amorphous (P-amor) phase is obtained, which can act as an intermediate state between the pure amorphous and pure crystalline phases for possible ternary-state data storage.Download high-res image (647KB)Download full-size image
Co-reporter:Mo-Ran Wang;Xiang-Yang Ren;Nian-Ke Chen;Hong-Bo Sun
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 38) pp:26164-26168
Publication Date(Web):2017/10/04
DOI:10.1039/C7CP05034H
The organic–inorganic hybrid perovskite has become a new type of semiconductor for low cost and highly efficient solar cells. However, the mechanism of interactions between the organic cation and the inorganic framework is still not completely clear under optical electronic excitation. In this work, we employ first-principles molecular dynamics with electronic excitation effects to prove that the hydrogen-bond interaction between the molecular cation and the inorganic lattice can be readily adjusted by several-percentage-valence-electron excitations in cubic CH3NH3PbI3. While the hydrogen-bond interaction causes serious lattice distortions, the electronic excitation can recover the lattice symmetry largely by weakening hydrogen bonding. The study offers atomic dynamics to understand the excitation process in the organic–inorganic hybrid perovskite semiconductor.
Co-reporter:Nian-Ke Chen;Dong Han;Feng Liu;Junhyeok Bang;Xue-Peng Wang;Qi-Dai Chen;Hai-Yu Wang;Shengbai Zhang;Hong-Bo Sun
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 36) pp:24735-24741
Publication Date(Web):2017/09/20
DOI:10.1039/C7CP03103C
Femtosecond lasers (fs) can cause a disparity between electronic and lattice temperatures in the very short period after irradiation. In this relatively cool lattice regime, the material properties can differ drastically from those under thermal equilibrium. In particular, first-principles calculations reveal two general mechanical effects on semiconductors. Firstly, the excitation can induce a negative pressure on the lattice, causing a >10% expansion, even for superhard diamond. Secondly, it induces inhomogeneous local forces on the atoms, for both perfect and distorted lattices. In the case of phase-change-memory for Ge2Sb2Te5 and GeTe alloys, such random forces cause a simultaneous phase transition from crystalline to amorphous, which enables faster data writing. These excitation effects are further supported by the time-dependent density functional theory. This work could be an important step in advancing fs laser techniques for the atomic-level control of structures, rather than relying on traditional melting or ablation approaches which often apply to much larger and non-atomic scales.
Co-reporter:Dan Wang;Hong-Bo Sun
Nanoscale (2009-Present) 2017 vol. 9(Issue 32) pp:11619-11624
Publication Date(Web):2017/08/17
DOI:10.1039/C7NR03389C
As a burgeoning two-dimensional (2D) semiconductor, InSe holds unique electronic properties and great promise for novel 2D electronic devices. To advance the development of 2D InSe devices, the exploration of n-type and p-type conductivities of InSe is indispensable. With first-principles calculations, we investigate the properties of native defects and substitutional impurities in monolayer InSe, including formation energies and ionization energies. As the traditional jellium scheme encounters an energy divergence for charged defects in 2D materials, an extrapolation approach is adopted here to obtain convergent energies. We find that In vacancy is a deep acceptor and Se vacancy is an electrically neutral defect. All the studied substitutional dopants at In or Se sites have high ionization energies in the range of 0.41–0.84 eV. However, electrons may transport through the defect-bound band edge states in XSe (X = Cl, Br, and I) as a potential source of n-type conductivity.
Co-reporter:Dan Wang, Xian-Bin Li, Dong Han, Wei Quan Tian, Hong-Bo Sun
Nano Today 2017 Volume 16(Volume 16) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.nantod.2017.07.001
•Importance of 2D semiconductors for nano/opto-electronics is discussed.•Defect atomic pictures are summarized via various experiments.•Two theoretical ways are used to evaluate electrical properties of defect.•Direct relations between defect and electronic device performance are reviewed.•A database of defects is summed up and the challenges are discussed.Two-dimensional (2D) semiconductors have attracted considerable attentions from electronic-engineering community due to their unique electronic properties. Especially, the inherent advantage of scaling semiconductor into atomic thickness has raised the prospect of possible extension of the Moore’s law. To achieve 2D electronics, a full comprehension of semiconductor defect physics and chemistry is indispensable due to its controlling electrical performance of 2D materials and functionalizing their devices. In this review, first we explain why 2D semiconductors is important for nanoelectronics and optoelectronics. Second, we elucidate how native defects or intentional impurities affect and control electrical characteristic in 2D semiconductors, such as carrier concentration and their conductive type. In this section, experimental pictures of defects and several updated theoretical methods to evaluate carrier ionization energies of defects and their conductive type are introduced in detail. Third, typical device experiments are shown to demonstrate a direct role of defects to functionalize 2D electronic device. Furthermore, a database of popular defects and their electrical properties in current popular 2D semiconductors is summarized for references. Last, we discuss the challenges and potential prospects of defect engineering for 2D devices. The present paper offers important viewpoints from semiconductor defects to design the emerging 2D electronics.Download high-res image (268KB)Download full-size image
Co-reporter:Sheng-Yi Xie, Xian-Bin Li, Wei Quan Tian, Nian-Ke Chen, Yeliang Wang, Shengbai Zhang and Hong-Bo Sun
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 2) pp:1093-1098
Publication Date(Web):07 Nov 2014
DOI:10.1039/C4CP03728F
Based on first-principles calculations, we designed for the first time a boron-kagome-based two-dimensional MgB6 crystal, in which two boron kagome layers sandwich a triangular magnesium layer. The two-dimensional lattice is metallic with several bands across the Fermi level, and among them a Dirac point appears at the K point of the first Brillouin zone. This metal-stabilized boron kagome system displays electron–phonon coupling, with a superconductivity critical transition temperature of 4.7 K, and thus it is another possible superconducting Mg–B compound besides MgB2. Furthermore, the proposed 2D MgB6 can also be used for hydrogen storage after decoration with Ca. Up to five H2 molecules can be attracted by one Ca with an average binding energy of 0.225 eV. The unique properties of 2D MgB6 will spur broad interest in nanoscience and technology.
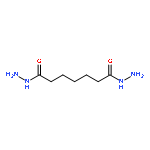
Co-reporter:Nian-Ke Chen, Xian-Bin Li, Xue-Peng Wang, Meng-Jiao Xia, Sheng-Yi Xie, Hai-Yu Wang, Zhitang Song, Shengbai Zhang, Hong-Bo Sun
Acta Materialia 2015 90() pp: 88-93
Publication Date(Web):
DOI:10.1016/j.actamat.2015.02.015
Co-reporter:Xilin Zhou, Mengjiao Xia, Feng Rao, Liangcai Wu, Xianbin Li, Zhitang Song, Songlin Feng, and Hongbo Sun
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 16) pp:14207
Publication Date(Web):August 4, 2014
DOI:10.1021/am503502q
Phase-change materials are highly promising for next-generation nonvolatile data storage technology. The pronounced effects of C doping on structural and electrical phase-change behaviors of Ge2Sb2Te5 material are investigated at the atomic level by combining experiments and ab initio molecular dynamics. C dopants are found to fundamentally affect the amorphous structure of Ge2Sb2Te5 by altering the local environments of Ge–Te tetrahedral units with stable C–C chains. The incorporated C increases the amorphous stability due to the enhanced covalent nature of the material with larger tetrahedral Ge sites. The four-membered rings with alternating atoms are reduced greatly with carbon addition, leading to sluggish phase transition and confined crystal grains. The lower RESET power is presented in the PCM cells with carbon-doped material, benefiting from its high resistivity and low thermal conductivity.Keywords: C doping; finite-element modeling; Ge2Sb2Te5; microstructure; molecular dynamic simulations; phase-change material
Co-reporter:Xue-Peng Wang, Nian-Ke Chen, Xian-Bin Li, Yan Cheng, X. Q. Liu, Meng-Jiao Xia, Z. T. Song, X. D. Han, S. B. Zhang and Hong-Bo Sun
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 22) pp:10810-10815
Publication Date(Web):24 Apr 2014
DOI:10.1039/C3CP55476G
The nano amorphous interface is important as it controls the phase transition for data storage. Yet, atomic scale insights into such kinds of systems are still rare. By first-principles calculations, we obtain the atomic interface between amorphous Si and amorphous Sb2Te3, which prevails in the series of Si–Sb–Te phase change materials. This interface model reproduces the experiment-consistent phenomena, i.e. the amorphous stability of Sb2Te3, which defines the data retention in phase change memory, and is greatly enhanced by the nano interface. More importantly, this method offers a direct platform to explore the intrinsic mechanism to understand the material function: (1) by steric effects through the atomic “channel” of the amorphous interface, the arrangement of the Te network is significantly distorted and is separated from the p-orbital bond angle in the conventional phase-change material; and (2) through the electronic “channel” of the amorphous interface, high localized electrons in the form of a lone pair are “projected” to Sb2Te3 from amorphous Si by a proximity effect. These factors set an effective barrier for crystallization and improve the amorphous stability, and thus data retention. The present research and scheme sheds new light on the engineering and manipulation of other key amorphous interfaces, such as Si3N4/Ge2Sb2Te5 and C/Sb2Te3, through first-principles calculations towards non-volatile phase change memory.
Co-reporter:Dong Han, Xian-Bin Li, Y.Y. Sun, S.B. Zhang, Sheng-Yi Xie, Sukit Limpijumnong, Zhan-Guo Chen, Hong-Bo Sun
Computational Materials Science 2014 Volume 82() pp:310-313
Publication Date(Web):1 February 2014
DOI:10.1016/j.commatsci.2013.09.065
•H prefers to reside in the hexagonal phase (hBN) rather than the cubic phase (cBN).•H defects including H2 molecular and H2∗∗ tend to gather to form cluster in hBN.•H can terminate a framework around impurity-induced sp3 nucleus in hBN.•In present of H, the growth of cBN can be suppressed.Hydrogen (H) behavior in crystal boron nitride (BN) has been systematically investigated by first-principles calculation. We find that H prefers to reside in the hexagonal phase (hBN) rather than the cubic phase (cBN). These kinds of H tend to gather to form clusters. In hBN, H can terminate a framework around an impurity-induced sp3 nucleus, thereby suppressing the cBN growth. This explains why there is no significant improvement in the hBN-to-cBN transition after aluminium (Al) doping.
Co-reporter:Sheng-Yi Xie;Dr. Xian-Bin Li; Wei Quan Tian;Dan Wang;Nian-Ke Chen;Dong Han; Hong-Bo Sun
ChemPhysChem 2014 Volume 15( Issue 13) pp:2707-2711
Publication Date(Web):
DOI:10.1002/cphc.201402057
Abstract
The reduction of graphene oxide can be used as a simple way to produce graphene on a large scale. However, the numerous edges produced by the oxidation of graphite seriously degrade the quality of the graphene and its carrier transport property. In this work, the reduction of oxygen-passivated graphene edges and the subsequent linking of separated graphene sheets by calcium are investigated by using first-principles calculations. The calculations show that calcium can effectively remove the oxygen groups from two adjacent edges. The joining point of the edges serves as the starting point of the reduction and facilitates the reaction. Once the oxygen groups are removed, the crack is sutured. If the joining point is lacking, it becomes difficult to zip the separated fragments. A general electron-reduction model and a random atom-reduction model are suggested for these two situations. The present study sheds light on the reduction of graphene-oxide edges by using reactive metals to give large-sized graphene through a simple chemical reaction.
Co-reporter:Sheng-Yi Xie, Xian-Bin Li, Y.Y. Sun, Yong-Lai Zhang, Dong Han, W.Q. Tian, Wen-Quan Wang, Yi-Song Zheng, S.B. Zhang, Hong-Bo Sun
Carbon 2013 Volume 52() pp:122-127
Publication Date(Web):February 2013
DOI:10.1016/j.carbon.2012.09.012
First-principles calculation identifies elementary processes in the thermal reduction of graphene oxide (GO) and reveals the effects of alkaline-earth metals (AEMs) in recovering the graphene. These metals are highly effective in removing residual oxygen groups resistive to thermal reduction, as well as healing the defects formed during the reduction, such as the carbonyl groups. In the AEM-assisted reduction, the AEMs serve as an electron reservoir of high chemical potential that forces electron transfer to the GO, whereas pristine carbon regions on the GO serve as a “bridge” to facilitate the electron transfer directly to oxidized carbon. This enables fast kinetics for the breaking of both C–O and CO bonds. Complete reduction is observed in our simulation at T ≤ 600 K within 32 ps for a 28%-oxygen-coverage GO model.
Co-reporter:Peng Sun, Lu You, Yanfeng Sun, Nianke Chen, Xianbin Li, Hongbo Sun, Jian Ma and Geyu Lu
CrystEngComm 2012 vol. 14(Issue 5) pp:1701-1708
Publication Date(Web):21 Dec 2011
DOI:10.1039/C1CE06197F
Novel Zn-doped SnO2hierarchical architectures were synthesized by a simple hydrothermal route. The observation of field-emission electron microscopy and transmission electron microscopy showed that Zn-doped SnO2 hierarchical architectures were composed of one-dimensional nanocones. Interestingly, these nanocones were almost parallel to each other and knitted by other parallel nanocones. The morphology of the products could be controlled by varying the concentration of Zn2+. A possible formation mechanism was proposed from the viewpoint of nucleation and the crystal growth habit. Evidences of dopant incorporation were demonstrated in the X-ray diffraction and X-ray photoelectron spectroscopy measurement of Zn-doped SnO2nanocones. The UV-vis absorption spectra of samples exhibited a blue shift with a decrease of the size of nanocones. Moreover, gas sensors based on the hierarchical Zn-doped SnO2nanocones displayed higher response to ethanol compared with the pure urchin-like SnO2 nanostructures. Finally, based on first-principles calculations, the enhancement in sensitivity toward ethanol could be explained by the strong coulomb binding between ZnSn and its neighboring O vacancies.
Co-reporter:Wei Liu, Fengchun Pang, Hailing Tu, Xianbin Li, Xiaoping Su, Shuyu Zhang, Chengsong Huo, Hai Yang
Journal of Alloys and Compounds 2012 Volume 545() pp:144-147
Publication Date(Web):25 December 2012
DOI:10.1016/j.jallcom.2012.08.022
First-principles simulation and PACVD experiment are used to reveal the atomic and electronic structure of BP4. With melting–quenching technique in molecule dynamics, for the first time we obtain the microscopic atomic picture for the amorphous BP4. Here, boron essentially retains a coordination number of 4 and sp3 electronic bonding whereas phosphorus has a lower coordination number of 3.2 and coexistence of sp3 and p electronic bonding. The cohesive energy of the present amorphous BP4 model is up to −5.32 eV/atom which is 1.6 times stronger than −3.37 eV/atom of the usual protected c-ZnS.Highlights► There is no investigation on the structure of the amorphous BPx film by calculation. ► We use MD combined with experiment to reveal the microstructure of amorphous BPx. ► Through MD, a typical representation for amorphous BP4 is obtained. ► For the first time we obtain the microscopic atomic picture for the amorphous BP4. ► B retain sp3 electronic bonding while P have coexistence of sp3 and p bonding.
Co-reporter:Xue-Peng Wang, Nian-Ke Chen, Xian-Bin Li, Yan Cheng, X. Q. Liu, Meng-Jiao Xia, Z. T. Song, X. D. Han, S. B. Zhang and Hong-Bo Sun
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 22) pp:NaN10815-10815
Publication Date(Web):2014/04/24
DOI:10.1039/C3CP55476G
The nano amorphous interface is important as it controls the phase transition for data storage. Yet, atomic scale insights into such kinds of systems are still rare. By first-principles calculations, we obtain the atomic interface between amorphous Si and amorphous Sb2Te3, which prevails in the series of Si–Sb–Te phase change materials. This interface model reproduces the experiment-consistent phenomena, i.e. the amorphous stability of Sb2Te3, which defines the data retention in phase change memory, and is greatly enhanced by the nano interface. More importantly, this method offers a direct platform to explore the intrinsic mechanism to understand the material function: (1) by steric effects through the atomic “channel” of the amorphous interface, the arrangement of the Te network is significantly distorted and is separated from the p-orbital bond angle in the conventional phase-change material; and (2) through the electronic “channel” of the amorphous interface, high localized electrons in the form of a lone pair are “projected” to Sb2Te3 from amorphous Si by a proximity effect. These factors set an effective barrier for crystallization and improve the amorphous stability, and thus data retention. The present research and scheme sheds new light on the engineering and manipulation of other key amorphous interfaces, such as Si3N4/Ge2Sb2Te5 and C/Sb2Te3, through first-principles calculations towards non-volatile phase change memory.
Co-reporter:Sheng-Yi Xie, Xian-Bin Li, Wei Quan Tian, Nian-Ke Chen, Yeliang Wang, Shengbai Zhang and Hong-Bo Sun
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 2) pp:NaN1098-1098
Publication Date(Web):2014/11/07
DOI:10.1039/C4CP03728F
Based on first-principles calculations, we designed for the first time a boron-kagome-based two-dimensional MgB6 crystal, in which two boron kagome layers sandwich a triangular magnesium layer. The two-dimensional lattice is metallic with several bands across the Fermi level, and among them a Dirac point appears at the K point of the first Brillouin zone. This metal-stabilized boron kagome system displays electron–phonon coupling, with a superconductivity critical transition temperature of 4.7 K, and thus it is another possible superconducting Mg–B compound besides MgB2. Furthermore, the proposed 2D MgB6 can also be used for hydrogen storage after decoration with Ca. Up to five H2 molecules can be attracted by one Ca with an average binding energy of 0.225 eV. The unique properties of 2D MgB6 will spur broad interest in nanoscience and technology.