Co-reporter:Houng Kang, Young Eun Lee, Peddiahgari Vasu Govardhana Reddy, Sangeeta Dey, Scott E. Allen, Kyle A. Niederer, Paul Sung, Kirsten Hewitt, Carilyn Torruellas, Madison R. Herling, and Marisa C. Kozlowski
Organic Letters October 20, 2017 Volume 19(Issue 20) pp:5505-5505
Publication Date(Web):October 12, 2017
DOI:10.1021/acs.orglett.7b02552
The first examples of asymmetric oxidative coupling of simple phenols and 2-hydroxycarbazoles are outlined. Generation of a more vanadium catalyst by ligand design and by addition of an exogenous Brønsted or Lewis acid was found to be key to coupling the more oxidatively resistant phenols. The resultant vanadium complex is both more Lewis acidic and more strongly oxidizing. Good to excellent levels of enantioselectivity could be obtained, and simple trituration readily provided the products with ≥95% ee.
Co-reporter:Osvaldo Gutierrez; John C. Tellis; David N. Primer; Gary A. Molander
Journal of the American Chemical Society 2015 Volume 137(Issue 15) pp:4896-4899
Publication Date(Web):April 2, 2015
DOI:10.1021/ja513079r
The cross-coupling of sp3-hybridized organoboron reagents via photoredox/nickel dual catalysis represents a new paradigm of reactivity for engaging alkylmetallic reagents in transition-metal-catalyzed processes. Reported here is an investigation into the mechanistic details of this important transformation using density functional theory. Calculations bring to light a new reaction pathway involving an alkylnickel(I) complex generated by addition of an alkyl radical to Ni(0) that is likely to operate simultaneously with the previously proposed mechanism. Analysis of the enantioselective variant of the transformation reveals an unexpected manifold for stereoinduction involving dynamic kinetic resolution (DKR) of a Ni(III) intermediate wherein the stereodetermining step is reductive elimination. Furthermore, calculations suggest that the DKR-based stereoinduction manifold may be responsible for stereoselectivity observed in numerous other stereoconvergent Ni-catalyzed cross-couplings and reductive couplings.
Co-reporter:Benedikt Wanner; Imants Kreituss; Osvaldo Gutierrez; Marisa C. Kozlowski;Jeffrey W. Bode
Journal of the American Chemical Society 2015 Volume 137(Issue 35) pp:11491-11497
Publication Date(Web):August 26, 2015
DOI:10.1021/jacs.5b07201
The catalytic kinetic resolution of cyclic amines with achiral N-heterocyclic carbenes and chiral hydroxamic acids has emerged as a promising method to obtain enantio-enriched amines with high selectivity factors. In this report, we describe the catalytic kinetic resolution of disubstituted piperdines with practical selectivity factors (s, up to 52) in which we uncovered an unexpected and pronounced conformational effect resulting in disparate reactivity and selectivity between the cis- and trans-substituted piperidine isomers. Detailed experimental and computational studies of the kinetic resolution of various disubstituted piperidines revealed a strong preference for the acylation of conformers in which the α-substituent occupies the axial position. This work provides further experimental and computational support for the concerted 7-member transition state model for acyl transfer reagents and expands the scope and functional group tolerance of the secondary amine kinetic resolution.
Co-reporter:Osvaldo Gutierrez, Dattatray Metil, Namrata Dwivedi, Nagaraju Gudimalla, E. R. R. Chandrashekar, Vilas H. Dahanukar, Apurba Bhattacharya, Rakeshwar Bandichhor, and Marisa C. Kozlowski
Organic Letters 2015 Volume 17(Issue 7) pp:1742-1745
Publication Date(Web):March 23, 2015
DOI:10.1021/acs.orglett.5b00520
The development of a practical and scalable process for the asymmetric synthesis of sitagliptin is reported. Density functional theory calculations reveal that two noncovalent interactions are responsible for the high diastereoselection. The first is an intramolecular hydrogen bond between the enamide NH and the boryl mesylate S═O, consistent with MsOH being crucial for high selectivity. The second is a novel C–H···F interaction between the aryl C5-fluoride and the methyl of the mesylate ligand.
Co-reporter:Lei Liu, Patrick J. Carroll, and Marisa C. Kozlowski
Organic Letters 2015 Volume 17(Issue 3) pp:508-511
Publication Date(Web):January 15, 2015
DOI:10.1021/ol503521b
The first regioselective oxidative coupling of 2-hydroxycarbazoles is described. With a vanadium catalyst and oxygen as the terminal oxidant, dimers with an ortho–ortho′ coupling pattern were obtained with high selectivity. Further oxidation led to ortho′–ortho′ coupling to generate a tetramer, which provided insight that the atropisomerization barriers of the unsymmetrical biaryl bonds are much lower than expected.
Co-reporter:Kelsey F. VanGelder and Marisa C. Kozlowski
Organic Letters 2015 Volume 17(Issue 23) pp:5748-5751
Publication Date(Web):November 20, 2015
DOI:10.1021/acs.orglett.5b02793
Catalytic conditions for the α-arylation of aryl nitromethanes have been discovered using parallel microscale experimentation, despite two prior reports of the lack of reactivity of these aryl nitromethane precursors. The method efficiently provides a variety of substituted, isolable diaryl nitromethanes. In addition, it is possible to sequentially append two different aryl groups to nitromethane. Mild oxidation conditions were identified to afford the corresponding benzophenones via the Nef reaction, and reduction conditions were optimized to afford several diaryl methylamines.
Co-reporter:Ryan R. Walvoord, Marisa C. Kozlowski
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3070-3074
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.12.105
Inexpensive and readily available cinchonidinium acetate is an effective catalyst for the syn-selective aza-Henry reaction of arylnitromethanes and aryl imines. The resulting masked cis-stilbenediamine products are produced in excellent diastereoselectivity and good enantioselectivity, and enantiopure material can be achieved via recrystallization. The features of the cinchona catalyst needed for selectivity are discussed, with specific emphasis on formation of a kinetically controlled syn-product without epimerization of the highly acidic α-nitro stereocenter.
Co-reporter:Alison E. Metz, Kailasham Ramalingam, Marisa C. Kozlowski
Tetrahedron Letters 2015 Volume 56(Issue 37) pp:5180-5184
Publication Date(Web):9 September 2015
DOI:10.1016/j.tetlet.2015.07.058
This study describes the synthesis of a class of anion-binding catalysts based on a xanthene scaffold. Both unsymmetrical catalysts and C2-symmetrical catalysts were generated, and were examined in the cyclization of 3- and 2-substituted furans onto N-acyliminium ions. Good conversion for each reaction was observed with a variety of anion-binding catalysts (42–76%).
Co-reporter:Alison E. Metz and Marisa C. Kozlowski
The Journal of Organic Chemistry 2015 Volume 80(Issue 1) pp:1-7
Publication Date(Web):November 28, 2014
DOI:10.1021/jo502408z
Because of their greater stability and unique conformational properties, unnatural amino acids are highly valued by pharmaceutical, biological, and organic chemists. This synopsis surveys the various catalytic methods used to access enantioenriched, acyclic α,α-disubstituted α-amino acids with a focus on the processes developed since 2007, when the last major reviews in this area were published.
Co-reporter:Kelsey F. VanGelder, Melinda Wang, and Marisa C. Kozlowski
The Journal of Organic Chemistry 2015 Volume 80(Issue 20) pp:10288-10293
Publication Date(Web):September 25, 2015
DOI:10.1021/acs.joc.5b01887
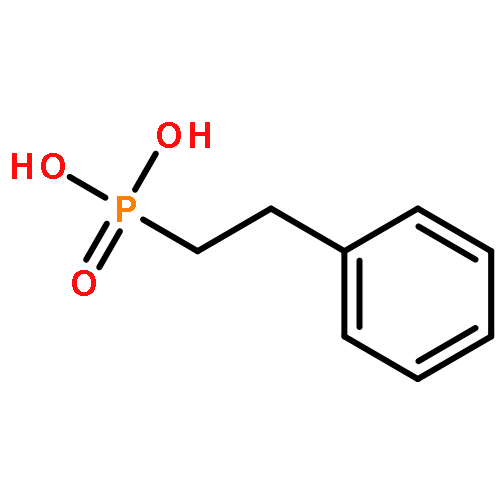
A versatile and general catalytic strategy has been developed for the α-arylation of phosphonoacetates utilizing parallel microscale experimentation. These α-substituted phosphonoacetates are widely useful, notably as substrates in the Horner–Wadsworth–Emmons-type olefinations. However, the current routes to these products involve harsh conditions, limiting the variety of functionality. The reported method can be used with a variety of aryl chlorides and aryl bromides, including several heterocyclic examples.
Co-reporter:John M. Curto
Journal of the American Chemical Society 2014 Volume 137(Issue 1) pp:18-21
Publication Date(Web):November 25, 2014
DOI:10.1021/ja5093166
The first selective coupling of a carbon nucleophile with methyl, ethyl, propyl, and butyl arenes in the absence of a directing group is described. Pd(OAc)2 double C–H activation displays remarkable selectivity for the terminal methyl sites in alkyl arenes, rather than the more commonly observed arene sp2 C–H activation. Mechanistic studies indicate the intermediacy of an azlactone dimer, obtained from oxidation with Pd(OAc)2, and are consistent with a Pd-catalyzed C–H activation vs a radical process. The observed reactivity establishes that typical reaction solvents (e.g., toluene) can readily participate in C–H activation chemistry.
Co-reporter:Alison E. Metz ; Erin E. Podlesny ; Patrick J. Carroll ; Ariel N. Klinghoffer
Journal of the American Chemical Society 2014 Volume 136(Issue 30) pp:10601-10604
Publication Date(Web):July 14, 2014
DOI:10.1021/ja506137j
An axial chiral tetrachlorinated bisbenzo[a]phenazine has been discovered that undergoes an alkane-induced shift in the solid state from a disordered amorphous form to an ordered polycrystalline form. This phase transition is caused by the formation of pores that accommodate linear alkanes of varying lengths with a very strong affinity as judged by differential scanning calorimetry. Single crystal X-ray structure analysis revealed that a series of weak phenolic OH···Cl hydrogen bonds dictates the pore structure. These weak interactions can be disrupted mechanically, causing the material to revert to the amorphous form. Notably, the interchange between the amorphous and crystalline forms is readily reversible and is easily observed by characteristic colorimetric changes. Measurements via photoimage processing reveal that the degree of color change is dictated by the type of alkane employed.
Co-reporter:Scott E. Allen ; Sheng-Ying Hsieh ; Osvaldo Gutierrez ; Jeffrey W. Bode
Journal of the American Chemical Society 2014 Volume 136(Issue 33) pp:11783-11791
Publication Date(Web):July 22, 2014
DOI:10.1021/ja505784w
The N-heterocyclic carbene and hydroxamic acid cocatalyzed kinetic resolution of cyclic amines generates enantioenriched amines and amides with selectivity factors up to 127. In this report, a quantum mechanical study of the reaction mechanism indicates that the selectivity-determining aminolysis step occurs via a novel concerted pathway in which the hydroxamic acid plays a key role in directing proton transfer from the incoming amine. This modality was found to be general in amide bond formation from a number of activated esters including those generated from HOBt and HOAt, reagents that are broadly used in peptide coupling. For the kinetic resolution, the proposed model accurately predicts the faster reacting enantiomer. A breakdown of the steric and electronic control elements shows that a gearing effect in the transition state is responsible for the observed selectivity.
Co-reporter:Young Eun Lee ; Trung Cao ; Carilyn Torruellas
Journal of the American Chemical Society 2014 Volume 136(Issue 19) pp:6782-6785
Publication Date(Web):May 5, 2014
DOI:10.1021/ja500183z
Simple catalysts that use atom-economical oxygen as the terminal oxidant to accomplish selective ortho–ortho, ortho–para, or para–para homo-couplings of phenols are described. In addition, chromium salen catalysts have been discovered as uniquely effective in the cross-coupling of different phenols with high chemo- and regioselectivity.
Co-reporter:Ryan R. Walvoord ; Phuong N. H. Huynh
Journal of the American Chemical Society 2014 Volume 136(Issue 45) pp:16055-16065
Publication Date(Web):October 17, 2014
DOI:10.1021/ja5086244
A spectrophotometric sensor is described that provides a useful assessment of the LUMO-lowering provided by catalysts in Diels–Alder and Friedel–Crafts reactions. A broad range of 33 hydrogen-bonding catalysts was assessed with the sensor, and the relative rates in the above reactions spanned 5 orders of magnitude as determined via 1H- and 2H NMR spectroscopic measurements, respectively. The differences between the maximum wavelength shift of the sensor with and without catalyst (Δλmax–1) were found to correlate linearly with ln(krel) values for both reactions, even though the substrate feature that interacts with the catalyst differs significantly (ketone vs nitro). The sensor provides an assessment of both the inherent reactivity of a catalyst architecture as well as the sensitivity of the reaction to changes within an architecture. In contrast, catalyst pKa values are a poor measure of reactivity, although correlations have been identified within catalyst classes.
Co-reporter:John M. Curto, Joshua S. Dickstein, Simon Berritt, and Marisa C. Kozlowski
Organic Letters 2014 Volume 16(Issue 7) pp:1948-1951
Publication Date(Web):March 25, 2014
DOI:10.1021/ol500506t
The first asymmetric synthesis of α-allyl-α-aryl α-amino acids by means of a three-component coupling of α-iminoesters, Grignard reagents, and cinnamyl acetate is reported. Notably, the enolate from the tandem process provides a much higher level of reactivity and selectivity than the same enolate generated via direct deprotonation, presumably due to differences in the solvation/aggregation state. A novel method for removal of a homoallylic amine protecting group delivers the free amine congeners. The α-allyl group offers a means to generate further valuable α-amino acid structures as exemplified by ring closing metathesis to generate a higher ring homologue of α-aryl-proline.
Co-reporter:Ludovic Raffier, Osvaldo Gutierrez, Gretchen R. Stanton, Marisa C. Kozlowski, and Patrick J. Walsh
Organometallics 2014 Volume 33(Issue 19) pp:5371-5377
Publication Date(Web):September 9, 2014
DOI:10.1021/om5007006
Alkenes have been discovered to be chelating groups to Zn(II), enforcing highly stereoselective additions of organozincs to β,γ-unsaturated ketones. 1H NMR studies and DFT calculations provide support for this surprising chelation mode. The results expand the range of coordinating groups for chelation-controlled carbonyl additions from heteroatom Lewis bases to simple C–C double bonds, broadening the 60 year old paradigm.
Co-reporter:John M. Curto and Marisa C. Kozlowski
The Journal of Organic Chemistry 2014 Volume 79(Issue 11) pp:5359-5364
Publication Date(Web):May 14, 2014
DOI:10.1021/jo500707t
Allylating agents were explored for the asymmetric synthesis of α-allyl-α-aryl α-amino acids by tandem N-alkylation/π-allylation. Cross-metathesis of the tandem product was developed to provide allylic diversity not afforded in the parent reaction; the synthesis of homotyrosine and homoglutamate analogues was completed. Cyclic α-amino acid derivatives could be accessed by ring-closing metathesis presenting a viable strategy to higher ring homologue of enantioenriched α-substituted proline. The eight-membered proline analogue was successfully converted to the pyrrolizidine natural product backbone.
Co-reporter:Scott E. Allen, Ryan R. Walvoord, Rosaura Padilla-Salinas, and Marisa C. Kozlowski
Chemical Reviews 2013 Volume 113(Issue 8) pp:6234
Publication Date(Web):June 20, 2013
DOI:10.1021/cr300527g
Co-reporter:Rosaura Padilla-Salinas, Ryan R. Walvoord, Sergei Tcyrulnikov, and Marisa C. Kozlowski
Organic Letters 2013 Volume 15(Issue 15) pp:3966-3969
Publication Date(Web):July 25, 2013
DOI:10.1021/ol401747u
A two-carbon homologation of vinyl triflates and bromides for the synthesis of homoallylic nitro products is described. This palladium-catalyzed double coupling of nitromethane exploits the anion stabilizing and leaving group properties of nitromethane, generating the homo allyl nitro products via a tandem cross-coupling/π-allylation sequence. The resultant process provides a mild and convenient entry to nitroethylated products, which are versatile precursors to β,γ-unsaturated carbonyls, homoallylic amines, and nitrile oxides.
Co-reporter:Alison E. Metz and Marisa C. Kozlowski
The Journal of Organic Chemistry 2013 Volume 78(Issue 2) pp:717-722
Publication Date(Web):December 17, 2012
DOI:10.1021/jo302071s
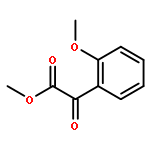
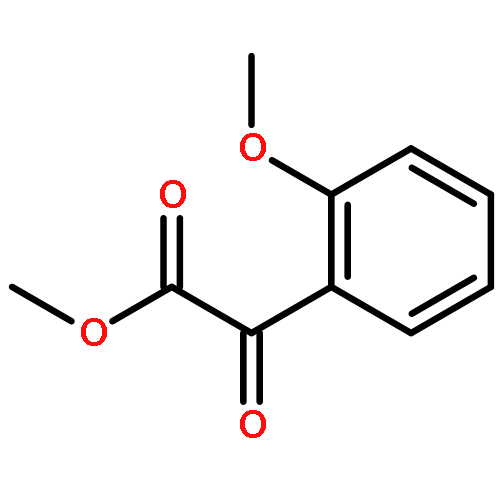
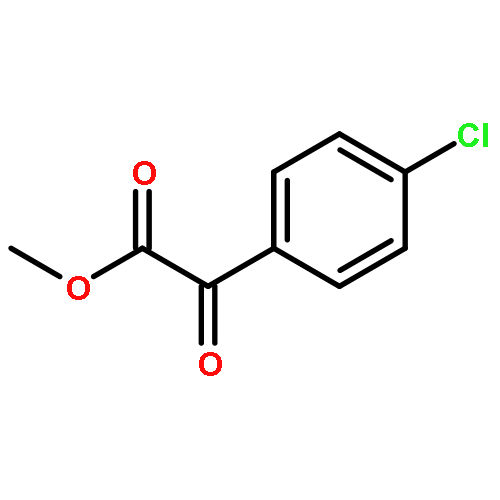
Nitroarylacetates are useful small molecular building blocks that act as precursors to α-ketoesters and aryl nitromethanes as well as α-amino acids. Methods were developed that produce each of these compound types in good yields. Two different conditions for decarboxylation are discussed for substrates with neutral and electron-poor aryl groups versus electron-rich aryl groups. For formation of the α-ketoesters, new mild conditions for the Nef disproportionation were identified.
Co-reporter:Erin E. Podlesny and Marisa C. Kozlowski
The Journal of Organic Chemistry 2013 Volume 78(Issue 2) pp:466-476
Publication Date(Web):December 18, 2012
DOI:10.1021/jo302364h
The development of the first asymmetric synthesis of a chiral anthraquinone dimer is outlined, resulting in the first total synthesis of (S)-bisoranjidiol. Rather than a biomimetic dimerization retrosynthetic disconnection, the anthracenyl ring systems are generated after formation of the axially chiral binaphthalene framework. This synthetic strategy has enabled the synthesis of several analogues. Key features of the synthesis include the enantioselective coupling of a hindered 2-naphthol containing substitution peri to the site of C–C bond formation, the regioselective oxidation of 8,8′-hydroxylated binaphthols to binaphtho-para-quinones, and a tandem regioselective Diels–Alder/aromatization reaction.
Co-reporter:Joshua S. Dickstein, John M. Curto, Osvaldo Gutierrez, Carol A. Mulrooney, and Marisa C. Kozlowski
The Journal of Organic Chemistry 2013 Volume 78(Issue 10) pp:4744-4761
Publication Date(Web):April 16, 2013
DOI:10.1021/jo400222c
Mechanism studies of a mild palladium-catalyzed decarboxylation of aromatic carboxylic acids are described. In particular, reaction orders and activation parameters for the two stages of the transformation were determined. These studies guided development of a catalytic system capable of turnover. Further evidence reinforces that the second stage, protonation of the arylpalladium intermediate, is the rate-determining step of the reaction. The first step, decarboxylative palladation, is proposed to occur through an intramolecular electrophilic palladation pathway, which is supported by computational and mechanism studies. In contrast to the reverse reaction (C–H insertion), the data support an electrophilic aromatic substitution mechanism involving a stepwise intramolecular protonation sequence for the protodepalladation portion of the reaction.
Co-reporter:Ryan R. Walvoord and Marisa C. Kozlowski
The Journal of Organic Chemistry 2013 Volume 78(Issue 17) pp:8859-8864
Publication Date(Web):July 30, 2013
DOI:10.1021/jo401249y
A method for the formation of arylnitromethanes is described that employs readily available aryl halides or triflates and small amounts of nitromethane in a dioxane solvent, thereby reducing the hazards associated with this reagent. Specifically, 2–10 equiv (1–5% v/v) of nitromethane can be employed in comparison to prior work that used nitromethane as solvent (185 equiv). The present transformation provides high yields at relatively low temperatures and tolerates an array of functionality, including heterocycles and substantial steric encumbrance.
Co-reporter:Phuong N. H. Huynh ; Ryan R. Walvoord
Journal of the American Chemical Society 2012 Volume 134(Issue 38) pp:15621-15623
Publication Date(Web):September 13, 2012
DOI:10.1021/ja3050663
A sensor has been developed to quickly and simply assess the relative reactivity of different hydrogen-bonding catalysts. Specifically, blue-shifts seen upon treatment of H-bonding catalysts with the colorimetric compound 7-methyl-2-phenylimidazo[1,2-a]pyrazin-3(7H)-one correlate well to the Keq of binding to the sensor. The blue-shifts also show a high degree of correlation with relative rates in Diels–Alder reactions of methyl vinyl ketone and cyclopentadiene employing the H-bonding catalysts. The relevance of the sensor blue-shifts to the LUMO-lowering abilities of the H-bonding catalysts is discussed.
Co-reporter:Scott E. Allen ; Jessada Mahatthananchai ; Jeffrey W. Bode
Journal of the American Chemical Society 2012 Volume 134(Issue 29) pp:12098-12103
Publication Date(Web):July 4, 2012
DOI:10.1021/ja302761d
The N-heterocyclic carbene catalyzed [4 + 2] cycloaddition has been shown to give γ,δ-unsaturated δ-lactones in excellent enantio- and diastereoselectivity. However, preliminary computational studies of the geometry of the intermediate enolate rendered ambiguous both the origins of selectivity and the reaction pathway. Here, we show that a concerted, but highly asynchronous, Diels–Alder reaction occurs rather than the stepwise Michael-type or Claisen-type pathways. In addition, two crucial interactions are identified that enable high selectivity: an oxyanion-steering mechanism and a CH−π interaction. The calculations accurately predict the enantioselectivity of a number of N-heterocyclic carbene catalysts in the hetero-Diels–Alder reaction.
Co-reporter:Ryan R. Walvoord, Simon Berritt, and Marisa C. Kozlowski
Organic Letters 2012 Volume 14(Issue 16) pp:4086-4089
Publication Date(Web):July 27, 2012
DOI:10.1021/ol301713j
An efficient cross-coupling reaction of aryl halides and nitromethane was developed with the use of parallel microscale experimentation. The arylnitromethane products are precursors for numerous useful synthetic products. An efficient method for their direct conversion to the corresponding oximes and aldehydes in a one-pot operation has been discovered. The process exploits inexpensive nitromethane as a carbonyl equivalent, providing a mild and convenient formylation method that is compatible with many functional groups.
Co-reporter:Erin E. Podlesny, Patrick J. Carroll, and Marisa C. Kozlowski
Organic Letters 2012 Volume 14(Issue 18) pp:4862-4865
Publication Date(Web):August 31, 2012
DOI:10.1021/ol302195e
The selective oxidation of a series of functionalized 8,8′-hydroxylated binaphthols to binaphtho-para- and binaphtho-ortho-quinones has been realized using either a Co-salen catalyst or ortho-iodoxybenzoic acid. A unique spirocyclic bis-spironaphthalenone was also obtained in good yield via a phenyliodonium diacetate promoted oxidative dearomatization.
Co-reporter:Alison E. Metz, Simon Berritt, Spencer D. Dreher, and Marisa C. Kozlowski
Organic Letters 2012 Volume 14(Issue 3) pp:760-763
Publication Date(Web):January 20, 2012
DOI:10.1021/ol203303b
Palladium-catalyzed cross-coupling conditions were developed that efficiently afford 2-aryl-2-nitroacetates from aryl bromides and the very acidic nitroacetates.
Co-reporter:Erin E. Podlesny
Journal of Natural Products 2012 Volume 75(Issue 6) pp:1125-1129
Publication Date(Web):June 12, 2012
DOI:10.1021/np300141t
The structure of a reported natural product isolate has been revised from (S)-2,2′-dimethoxy-[1,1′-binaphthalene]-5,5′,6,6′-tetraol to a known tetrabrominated diphenyl ether. After total synthesis of the reported binaphthalenetetrol was accomplished via a key reduction of a binaphtho-ortho-quinone, comparison of the physical properties and NMR spectroscopic data of the synthetic material indicated that the structure of the natural product isolate was incorrect. Evaluation of the authentic natural product suggested the structure is a tetrabrominated diphenyl ether, likely 3,5-dibromo-2-(3,5-dibromo-2-methoxyphenoxy)phenol.
Co-reporter:Carol A. Mulrooney;Erin M. O'Brien;Barbara J. Morgan
European Journal of Organic Chemistry 2012 Volume 2012( Issue 21) pp:3887-3904
Publication Date(Web):
DOI:10.1002/ejoc.201200184
Abstract
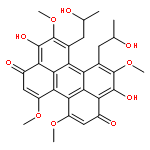
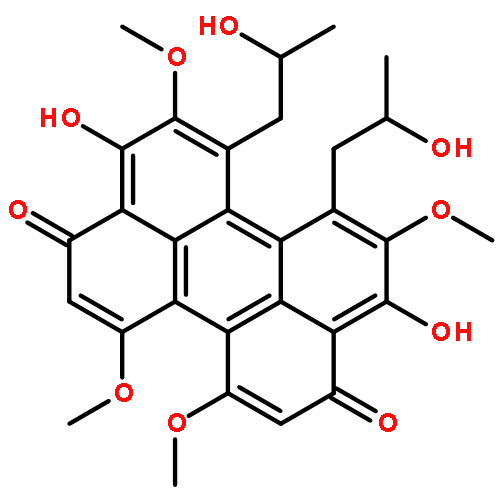
The perylenequinones are a class of natural products characterized by a pentacyclic conjugated chromophore giving rise to photoactivity. Potentially useful light-activated biological activity, targeting protein kinase C (PKC), has been identified for several of the natural products. Recently discovered new members of this compound class, as well as several related phenanthro-perylenequinones, are reviewed. Natural product modifications that improve biological profiles are outlined, as well as avenues for the total synthesis of analogs not available from the natural product series. An overview of structure/function relationships is provided.
Co-reporter:Trung Cao, Elizabeth C. Linton, Joshua Deitch, Simon Berritt, and Marisa C. Kozlowski
The Journal of Organic Chemistry 2012 Volume 77(Issue 24) pp:11034-11055
Publication Date(Web):November 20, 2012
DOI:10.1021/jo302039n
In this Article, a strategy to obtain highly enantioselective catalysts for the Claisen rearrangement of allyloxy- and propargyloxy-indoles is outlined. Ultimately, copper BOX and palladium BINAP or PHOX catalysts were discovered as superior in catalyzing Claisen rearrangements of allyloxy- or proparyloxy-substituted indoles to generate oxindoles bearing allyl- or allenyl-substituted quaternary centers. This method proved to be tolerant of a broad range of functional groups. Tandem reactions of the silyl-allene products provide rapid access to a variety of spirocyclic oxindoles in one operation.
Co-reporter:Trung Cao;Joshua Deitch;Dr. Elizabeth C. Linton ; Marisa C. Kozlowski
Angewandte Chemie 2012 Volume 124( Issue 10) pp:2498-2501
Publication Date(Web):
DOI:10.1002/ange.201107417
Co-reporter:Trung Cao;Joshua Deitch;Dr. Elizabeth C. Linton ; Marisa C. Kozlowski
Angewandte Chemie International Edition 2012 Volume 51( Issue 10) pp:2448-2451
Publication Date(Web):
DOI:10.1002/anie.201107417
Co-reporter:Rakeshwar Bandichhor, Andrew N. Lowell, and Marisa C. Kozlowski
The Journal of Organic Chemistry 2011 Volume 76(Issue 16) pp:6475-6487
Publication Date(Web):June 27, 2011
DOI:10.1021/jo200398v
Two methods are presented that were designed to circumvent the persistent problem of benzofuran formation and instead yield a spiroketal of the rubromycin family type. First, using an alternative disconnection, a hemiketal conjugate addition to a naphthaquinone electrophile was investigated. Synthesis of the requisite electrophile provided insight into the selective oxidation and functionalization of the naphthalene portion. Second, the electronic features of the isocoumarin ring system were adjusted, and the corresponding reactivity further supports the hypothesis that electron-rich isocoumarins are capable of spiroketalization. Robust, flexible syntheses from simple precursors were developed that allowed multiple reduced isocoumarins to be generated. Combined, the data presented herein give insight into the sensitivities of this family and illuminate other potential methods of spiroketalization. In addition, the convergent assembly of substrates containing different naphthaquinone and isocoumarin subunits highlights the utility of our 1,3-dipolar cycloaddition approach to generate analogs of these structures for SAR, as well as chemical reactivity studies.
Co-reporter:Andrew N. Lowell, Michael W. Fennie, and Marisa C. Kozlowski
The Journal of Organic Chemistry 2011 Volume 76(Issue 16) pp:6488-6502
Publication Date(Web):June 27, 2011
DOI:10.1021/jo200399z
The central portion of purpuromycin has been assembled via a classical spiroketalization reaction. Key to promoting this reaction mode versus benzofuran formation was the oxidation state of the spiroketal core. With a higher oxidation state, even the electron-deficient isocoumarin found in purpuromycin could be employed directly in the spiroketalization. The two halves of the spiroketalization precursor were joined via a nitrile oxide/styrene 1,3-dipolar cycloaddition. A very mild selenium dioxide oxidation was used to introduce the required oxidation state of the spiroketal core.
Co-reporter:Andrew N. Lowell, Philip D. Wall, Stephen P. Waters, Marisa C. Kozlowski
Tetrahedron 2010 66(30) pp: 5573-5582
Publication Date(Web):
DOI:10.1016/j.tet.2010.05.077
Co-reporter:Marisa C. Kozlowski, James C. Ianni
Journal of Molecular Catalysis A: Chemical 2010 324(1–2) pp: 141-145
Publication Date(Web):
DOI:10.1016/j.molcata.2010.03.030
Co-reporter:Barbara J. Morgan, Carol A. Mulrooney and Marisa C. Kozlowski
The Journal of Organic Chemistry 2010 Volume 75(Issue 1) pp:44-56
Publication Date(Web):November 9, 2009
DOI:10.1021/jo9013854
The evolution of the first total synthesis of perylenequinone cercosporin is described. The key features developed during these efforts include a biscuprate epoxide alkylation, installation of the methylidene acetal, palladium-catalyzed O-arylation, and C3,C3′-decarbonylation. Due to the rapid atropisomerization of the helical axis of cercosporin (at 37 °C), the sequencing of these transformations was critical. To this end, the developed protocol enabled the formation of a key advanced intermediate on preparative scale absent any atropisomerization. Furthermore, the O-arylation proved to be general, and the strategy was used in an improved synthesis of a helical chiral perylenequinone structure.
Co-reporter:Marisa C. Kozlowski, Barbara J. Morgan and Elizabeth C. Linton
Chemical Society Reviews 2009 vol. 38(Issue 11) pp:3193-3207
Publication Date(Web):23 Sep 2009
DOI:10.1039/B821092F
This tutorial review highlights the use of catalytic asymmetric 2-naphthol couplings in total synthesis. The types of chirality, chiral biaryl natural products, prior approaches to chiral biaryl natural products, and other catalytic asymmetric biaryl couplings are outlined. The three main categories of chiral catalysts for 2-naphthol coupling (Cu, V, Fe) are described with discussion of their limitations and advantages. Applications of the copper catalyzed couplings in biomimetic syntheses are discussed including nigerone, hypocrellin, calphostin D, phleichrome, and cercosporin.
Co-reporter:Barbara J. Morgan ; Sangeeta Dey ; Steven W. Johnson
Journal of the American Chemical Society 2009 Volume 131(Issue 26) pp:9413-9425
Publication Date(Web):June 2, 2009
DOI:10.1021/ja902324j
The total syntheses of the PKC inhibitors (+)-calphostin D, (+)-phleichrome, cercosporin, and 10 novel perylenequinones are detailed. The highly convergent and flexible strategy developed employed an enantioselective oxidative biaryl coupling and a double cuprate epoxide opening, allowing the selective syntheses of all the possible stereoisomers in pure form. In addition, this strategy permitted rapid access to a broad range of analogues, including those not accessible from the natural products. These compounds provided a powerful means for evaluation of the perylenequinone structural features necessary to PKC activity. Simpler analogues were discovered with superior PKC inhibitory properties and superior photopotentiation in cancer cell lines relative to the more complex natural products.
Co-reporter:MichaelT. Wentzel;J. Brian Hewgley;RajeshM. Kamble;PhilipD. Wall ;MarisaC. Kozlowski
Advanced Synthesis & Catalysis 2009 Volume 351( Issue 6) pp:931-937
Publication Date(Web):
DOI:10.1002/adsc.200800730
Co-reporter:Joshua S. Dickstein and Marisa C. Kozlowski
Chemical Society Reviews 2008 vol. 37(Issue 6) pp:1166-1173
Publication Date(Web):01 Apr 2008
DOI:10.1039/B709139G
α-Iminoesters are useful precursors to substituted α-amino acid derivatives and are utilized in a number of organic transformations. As a consequence of the adjacent ester functionality, these imines are more reactive relative to other types of imines. While a significant body of work has focused on nucleophilic additions to the imine carbon (C-alkylation), a second pathway that involves nucleophilic reaction at the nitrogen (N-alkylation) is much less explored. This tutorial review highlights work that has exploited this unusual α-iminoester reactivity mode.
Co-reporter:Marisa C. Kozlowski;Elizabeth C. Dugan;Evan S. DiVirgilio;Katja Maksimenka;Gerhard Bringmann
Advanced Synthesis & Catalysis 2007 Volume 349(Issue 4-5) pp:
Publication Date(Web):20 MAR 2007
DOI:10.1002/adsc.200600570
An enantioselective synthesis of the chiral bisnaphthopyrone natural product nigerone and its enantiomer, ent-nigerone, has been realized. The use of constrained 2-naphthol substrates was critical to producing highly functionalized chiral 1,1′-binaphthols via asymmetric oxidative biaryl coupling with 1,5-diaza-cis-decalin copper complexes. The final natural product was formed via a key eight-step isomerization process of the coupling product, bisisonigerone, and proceeded with retention of the biaryl configuration. The axial configurations of bisisonigerone and nigerone were definitively established by a combination of circular dichroism (CD) measurements and quantum chemical CD calculations.
Co-reporter:James C. Ianni Dr.;Venkatachalam Annamalai;Puay-Wah Phuan Dr.;Manoranjan Pa Dr. and
Angewandte Chemie 2006 Volume 118(Issue 33) pp:
Publication Date(Web):11 AUG 2006
DOI:10.1002/ange.200600329
Exzellente Vorhersage: Die Selektivitäten eines Satzes chiraler Katalysatoren wurden mit Methoden vorhergesagt, die von quantenmechanischen Feldern für molekulare Wechselwirkungen abgeleitet sind und auf Grundzustands- statt auf Übergangszustandsstrukturen angewendet wurden. Die Vorhersagen für die asymmetrische Addition von Et2Zn an PhCHO stimmen bemerkenswert gut mit den experimentellen Daten überein (=0.87).
Co-reporter:James C. Ianni Dr.;Venkatachalam Annamalai;Puay-Wah Phuan Dr.;Manoranjan Pa Dr. and
Angewandte Chemie International Edition 2006 Volume 45(Issue 33) pp:
Publication Date(Web):11 AUG 2006
DOI:10.1002/anie.200600329
Excellent forecast: The selectivities for a set of chiral catalysts were predicted by methods derived from quantum mechanical molecular interaction fields that were applied to ground-state structures rather than transition-state structures. The predictions for the asymmetric addition of Et2Zn to PhCHO are in remarkable agreement with the experimental results (=0.87).
Co-reporter:Marisa C. Kozlowski, Evan S. DiVirgilio, Krishnan Malolanarasimhan, Carol A. Mulrooney
Tetrahedron: Asymmetry 2005 Volume 16(Issue 21) pp:3599-3605
Publication Date(Web):31 October 2005
DOI:10.1016/j.tetasy.2005.10.008
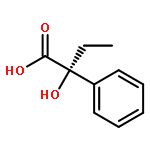
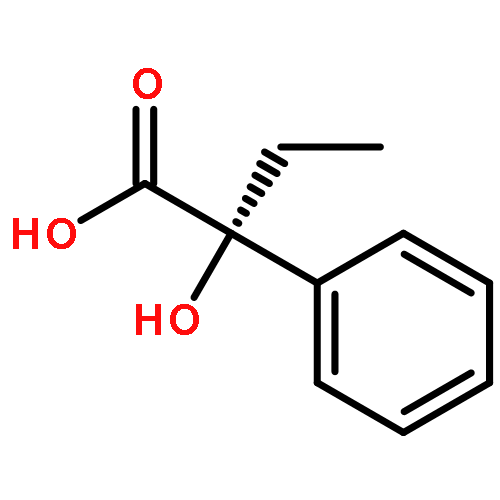
The menthol esters of α-phenylacetate derivatives undergo diastereoselective oxidative dimerizations (α-cyano) in the presence of oxidants, and nitrosylation (α-amido) in the presence of CAN to form products containing adjacent functionalized quaternary stereocenters and tertiary alcohols, respectively.(2R,3S)-Bis(2-isopropyl-5-methylcyclohexyl) 2,3-dicyano-2,3-diphenylsuccinateC38H48N2O4[α]D23=-45.1 (c 0.61, CHCl3)(2R,3S)-Bis(2-isopropyl-5-methylcyclohexyl) 2,3-dicyano-2,3-dip-tolylsuccinateC40H52N2O4[α]D23=-50.2 (c 0.96, CHCl3)(2S,3S)-Bis(2-isopropyl-5-methylcyclohexyl) 2,3-dicyano-2,3-dip-tolylsuccinateC40H52N2O4[α]D23=-148.5 (c 0.90, CHCl3)(2S)-2-Isopropyl-5-methylcyclohexyl 3-amino-2-hydroxy-3-oxo-2-phenylpropanoateC19H27NO4[α]D23=-88.7 (c 0.62, CHCl3)(5S)-2-Isopropyl-5-methylcyclohexyl 2,2-dimethyl-4-oxo-5-phenyloxazolidine-5-carboxylateC22H31NO4[α]D23=-53.4 (c 0.72, CHCl3)(R)-2,3-Dihydroxy-N-isopropyl-2-phenylpropanamideC12H17NO3[α]D23=+3.5 (c 0.38, CHCl3)
Co-reporter:Xu Xie;Puay-Wah Phuan
Angewandte Chemie 2003 Volume 115(Issue 19) pp:
Publication Date(Web):14 MAY 2003
DOI:10.1002/ange.200250325
Einfach, aber gut geordnet! Durch eine Folge von zwei unterschiedlichen Kupplungreaktionen können aus Alkinylnaphtholen mit hoher Stereoselektivität chirale Binaphthyl-Polymere erhalten werden (siehe Bild). Die bemerkenswerte Chemoselektivität der Kupplungsschritte ermöglicht den gezielten Aufbau funktionalisierter Produkte in einer Eintopfreaktion.
Co-reporter:Xu Xie;Puay-Wah Phuan
Angewandte Chemie International Edition 2003 Volume 42(Issue 19) pp:
Publication Date(Web):14 MAY 2003
DOI:10.1002/anie.200250325
Unusually orderly! Alkynylnaphthols undergo two kinds of couplings stepwise, with high stereocontrol, and in one pot to give chiral binaphthyl polymers like that shown. Since the chemoselectivity of each coupling is remarkably high, this approach is useful for the organized assembly of multifunctional substrates in a single operation.
Co-reporter:Manoranjan Panda, Puay-Wah Phuan and Marisa C. Kozlowski
Chemical Communications 2002 (Issue 14) pp:1552-1553
Publication Date(Web):19 Jun 2002
DOI:10.1039/B201950G
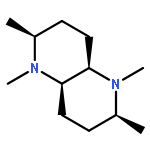
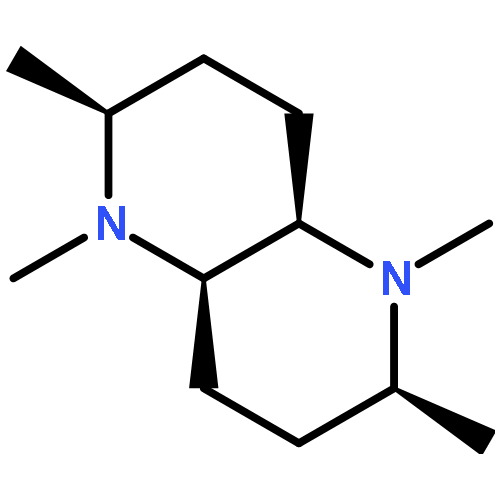
Calculated 13C NMR chemical shifts were key to assigning the structures of the conformational forms of complexed and uncomplexed bispidine derivatives
Co-reporter:Erin E. Podlesny
Organic Letters () pp:
Publication Date(Web):February 24, 2012
DOI:10.1021/ol3001365
The first enantioselective total synthesis of the bisanthraquinone (S)-bisoranjidiol and an unnatural regioisomer has been accomplished. Key features of the synthesis include the asymmetric oxidative biaryl coupling of a hindered 8-substituted 2-naphthol, selective para-quinone formation, and regioselective tandem Diels–Alder/aromatization reactions.
Co-reporter:Joshua S. Dickstein and Marisa C. Kozlowski
Chemical Society Reviews 2008 - vol. 37(Issue 6) pp:NaN1173-1173
Publication Date(Web):2008/04/01
DOI:10.1039/B709139G
α-Iminoesters are useful precursors to substituted α-amino acid derivatives and are utilized in a number of organic transformations. As a consequence of the adjacent ester functionality, these imines are more reactive relative to other types of imines. While a significant body of work has focused on nucleophilic additions to the imine carbon (C-alkylation), a second pathway that involves nucleophilic reaction at the nitrogen (N-alkylation) is much less explored. This tutorial review highlights work that has exploited this unusual α-iminoester reactivity mode.
Co-reporter:Marisa C. Kozlowski, Barbara J. Morgan and Elizabeth C. Linton
Chemical Society Reviews 2009 - vol. 38(Issue 11) pp:NaN3207-3207
Publication Date(Web):2009/09/23
DOI:10.1039/B821092F
This tutorial review highlights the use of catalytic asymmetric 2-naphthol couplings in total synthesis. The types of chirality, chiral biaryl natural products, prior approaches to chiral biaryl natural products, and other catalytic asymmetric biaryl couplings are outlined. The three main categories of chiral catalysts for 2-naphthol coupling (Cu, V, Fe) are described with discussion of their limitations and advantages. Applications of the copper catalyzed couplings in biomimetic syntheses are discussed including nigerone, hypocrellin, calphostin D, phleichrome, and cercosporin.
.jpg)





![[1,1'-Biphenyl]-4,4'-diol, 2,2'-bis(1,1-dimethylethyl)-5,5'-dimethyl-](http://img.cochemist.com/ccimg/10400/10378-11-7.png)
![[1,1'-Biphenyl]-4,4'-diol, 2,2'-bis(1,1-dimethylethyl)-5,5'-dimethyl-](http://img.cochemist.com/ccimg/10400/10378-11-7_b.png)












-](http://img.cochemist.com/ccimg/5900/5809-26-7.png)
-](http://img.cochemist.com/ccimg/5900/5809-26-7_b.png)





![6-phenyl-7-oxabicyclo[4.1.0]heptane](http://img.cochemist.com/ccimg/4900/4829-01-0.png)
![6-phenyl-7-oxabicyclo[4.1.0]heptane](http://img.cochemist.com/ccimg/4900/4829-01-0_b.png)



![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6.png)
![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6_b.png)

![(2E)-2-CYANO-N-[2-(1H-INDOL-3-YL)ETHYL]-3-[4-(METHYLSULFANYL)PHEN<WBR />YL]ACRYLAMIDE](http://img.cochemist.com/ccimg/3700/3692-67-9.png)
![(2E)-2-CYANO-N-[2-(1H-INDOL-3-YL)ETHYL]-3-[4-(METHYLSULFANYL)PHEN<WBR />YL]ACRYLAMIDE](http://img.cochemist.com/ccimg/3700/3692-67-9_b.png)
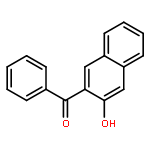
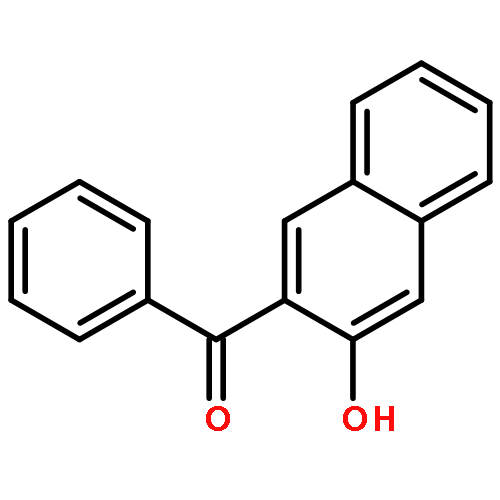
![Pyrrolidine, 1-[(3-hydroxy-2-naphthalenyl)carbonyl]-](http://img.cochemist.com/ccimg/3700/3665-55-2.png)
![Pyrrolidine, 1-[(3-hydroxy-2-naphthalenyl)carbonyl]-](http://img.cochemist.com/ccimg/3700/3665-55-2_b.png)



![[1,1'-Biphenyl]-2-ol,2,2'',2''''-phosphite](http://img.cochemist.com/ccimg/2800/2752-19-4.png)
![[1,1'-Biphenyl]-2-ol,2,2'',2''''-phosphite](http://img.cochemist.com/ccimg/2800/2752-19-4_b.png)



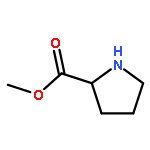
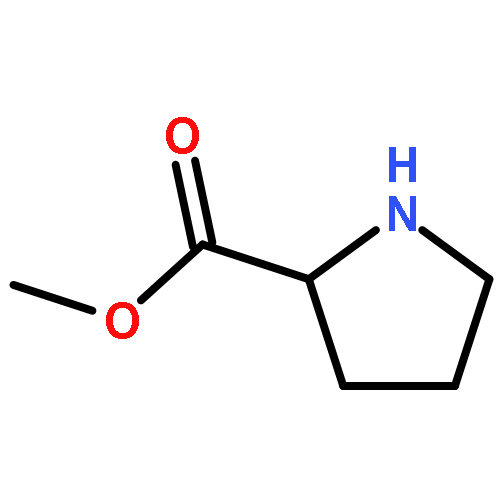











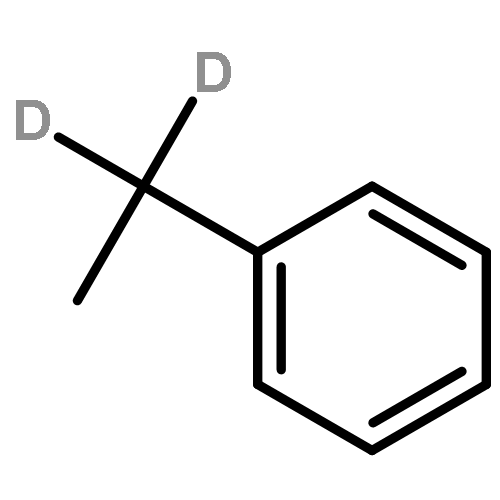
![Urea, N-[3,5-bis(trifluoromethyl)phenyl]-N'-phenyl-](http://img.cochemist.com/ccimg/1700/1644-96-8.png)
![Urea, N-[3,5-bis(trifluoromethyl)phenyl]-N'-phenyl-](http://img.cochemist.com/ccimg/1700/1644-96-8_b.png)






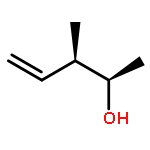







![Benzene, [(1E)-2-bromoethenyl]-](/data/chemimg/1476700/588-72-7.png)
![Benzene, [(1E)-2-bromoethenyl]-](/data/chemimg/1476700/588-72-7_b.png)




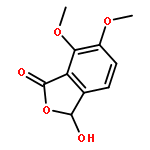
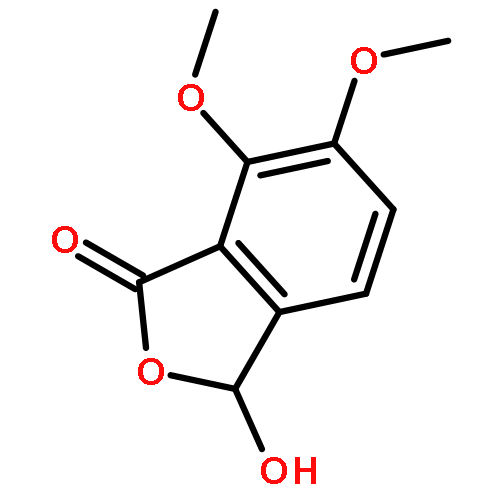
![3,3',5,5'-Tetra-tert-butyl-[1,1'-biphenyl]-4,4'-diol](http://img.cochemist.com/ccimg/200/128-38-1.png)
![3,3',5,5'-Tetra-tert-butyl-[1,1'-biphenyl]-4,4'-diol](http://img.cochemist.com/ccimg/200/128-38-1_b.png)
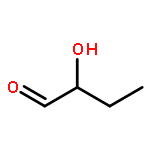
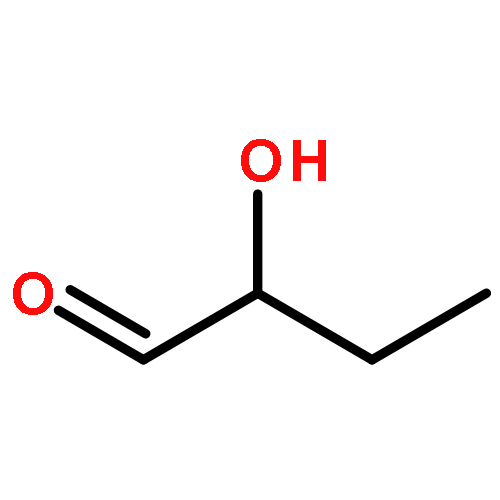
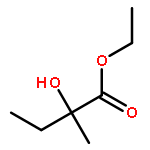



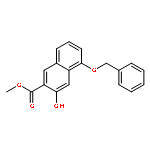
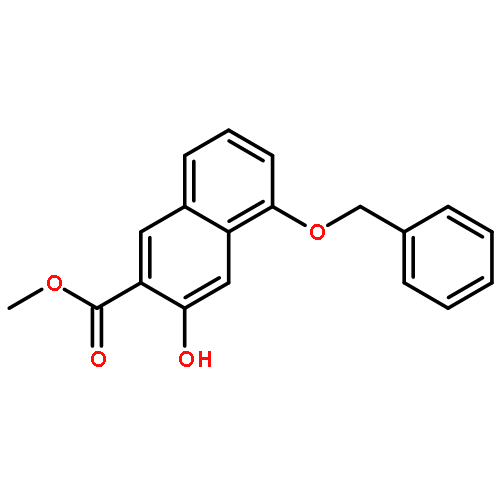
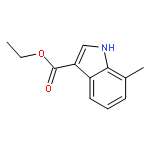
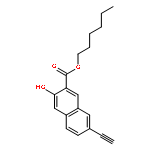
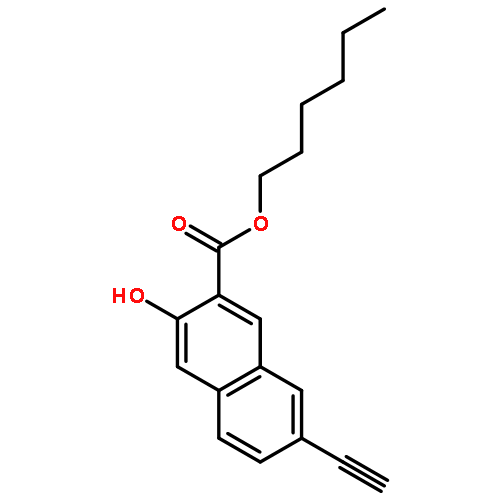
![POLY[3,3'-BIS[(HEXYLOXY)CARBONYL]-2,2'-DIHYDROXY[1,1'-BINAPHTHALENE]-6,6'-DIYL]](http://img.cochemist.com/ccimg/577100/577040-00-7.png)
![POLY[3,3'-BIS[(HEXYLOXY)CARBONYL]-2,2'-DIHYDROXY[1,1'-BINAPHTHALENE]-6,6'-DIYL]](http://img.cochemist.com/ccimg/577100/577040-00-7_b.png)
![2-Naphthalenol, 7-methoxy-3-[(4-methoxyphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/574100/574001-89-1.png)
![2-Naphthalenol, 7-methoxy-3-[(4-methoxyphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/574100/574001-89-1_b.png)
![2-Naphthalenol, 3-[(4-methoxyphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/574100/574001-86-8.png)
![2-Naphthalenol, 3-[(4-methoxyphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/574100/574001-86-8_b.png)
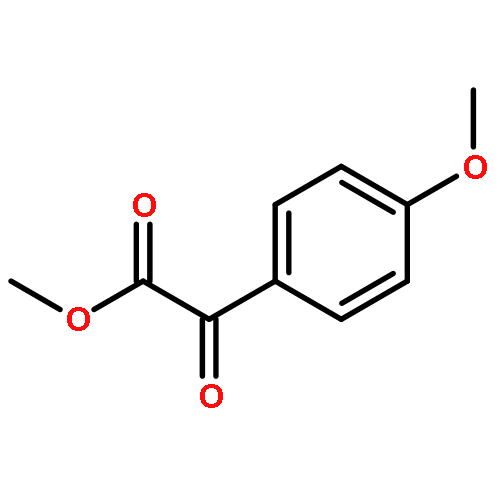
![Propanedioic acid, [(4-methoxyphenyl)acetyl]-, dimethyl ester](http://img.cochemist.com/ccimg/567000/566933-79-7.png)
![Propanedioic acid, [(4-methoxyphenyl)acetyl]-, dimethyl ester](http://img.cochemist.com/ccimg/567000/566933-79-7_b.png)
![Propanedioic acid, [(3,4,5-trimethoxyphenyl)acetyl]-, dimethyl ester](http://img.cochemist.com/ccimg/567000/566933-77-5.png)
![Propanedioic acid, [(3,4,5-trimethoxyphenyl)acetyl]-, dimethyl ester](http://img.cochemist.com/ccimg/567000/566933-77-5_b.png)






![Propanedioic acid, [(3,5-dimethoxyphenyl)acetyl]-, dimethyl ester](http://img.cochemist.com/ccimg/468100/468078-54-8.png)
![Propanedioic acid, [(3,5-dimethoxyphenyl)acetyl]-, dimethyl ester](http://img.cochemist.com/ccimg/468100/468078-54-8_b.png)

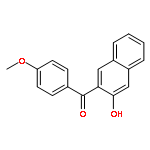

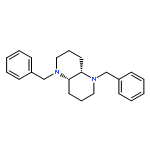
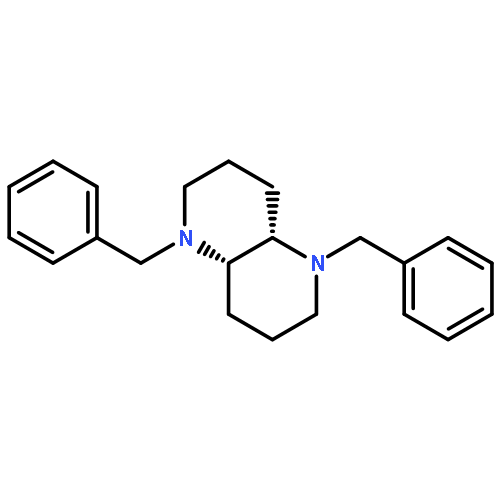
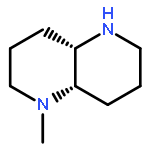
![Bicyclo[2.2.1]heptan-2-ol,1,7,7-trimethyl-3-(4-morpholinyl)-, (1R,2S,3R,4S)-](http://img.cochemist.com/ccimg/287200/287105-48-0.png)
![Bicyclo[2.2.1]heptan-2-ol,1,7,7-trimethyl-3-(4-morpholinyl)-, (1R,2S,3R,4S)-](http://img.cochemist.com/ccimg/287200/287105-48-0_b.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 3-phenyl-, (1S,2S,3S,4R)-](http://img.cochemist.com/ccimg/279700/279685-68-6.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 3-phenyl-, (1S,2S,3S,4R)-](http://img.cochemist.com/ccimg/279700/279685-68-6_b.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 3-methyl-, (1S,2R,3S,4R)-](http://img.cochemist.com/ccimg/278200/278173-24-3.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde, 3-methyl-, (1S,2R,3S,4R)-](http://img.cochemist.com/ccimg/278200/278173-24-3_b.png)

![Imidazo[1,2-a]pyrazin-3(7H)-one, 7-methyl-2-phenyl-](http://img.cochemist.com/ccimg/152800/152719-99-8.png)
![Imidazo[1,2-a]pyrazin-3(7H)-one, 7-methyl-2-phenyl-](http://img.cochemist.com/ccimg/152800/152719-99-8_b.png)

![3-[tert-butyl(dimethyl)silyl]prop-2-yn-1-ol](http://img.cochemist.com/ccimg/120800/120789-51-7.png)
![3-[tert-butyl(dimethyl)silyl]prop-2-yn-1-ol](http://img.cochemist.com/ccimg/120800/120789-51-7_b.png)






















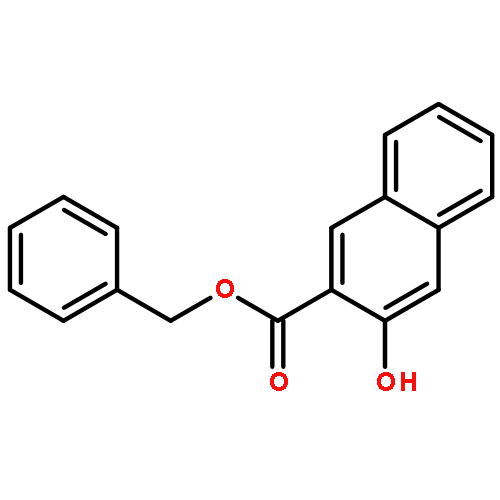
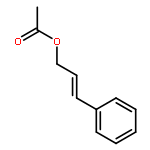
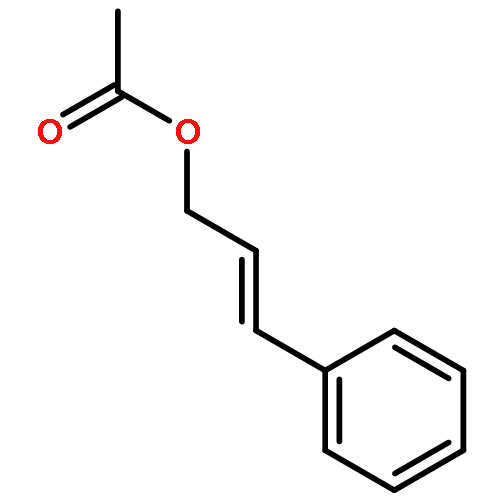


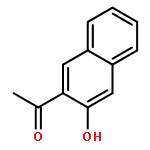
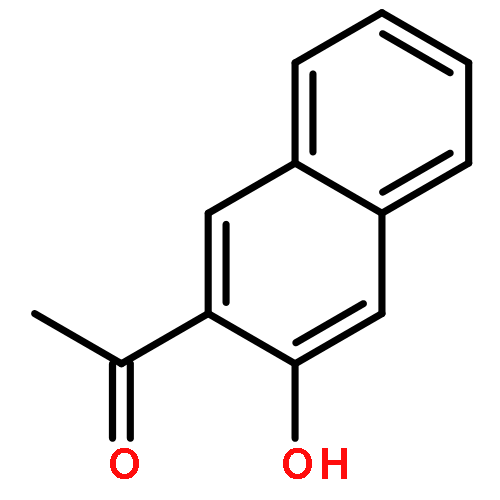
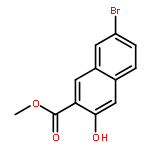
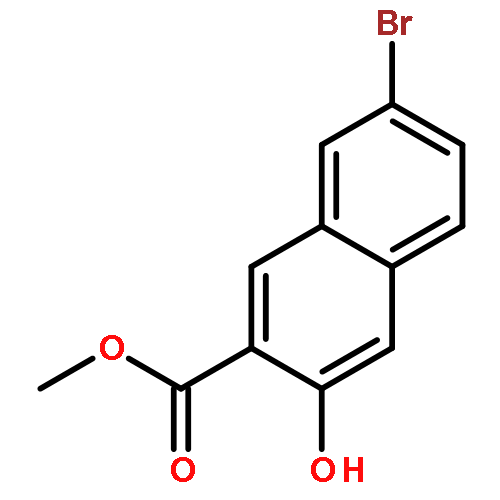






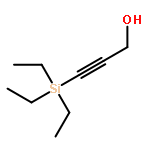
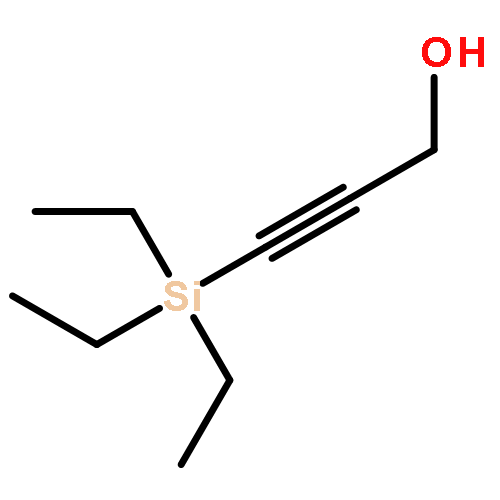

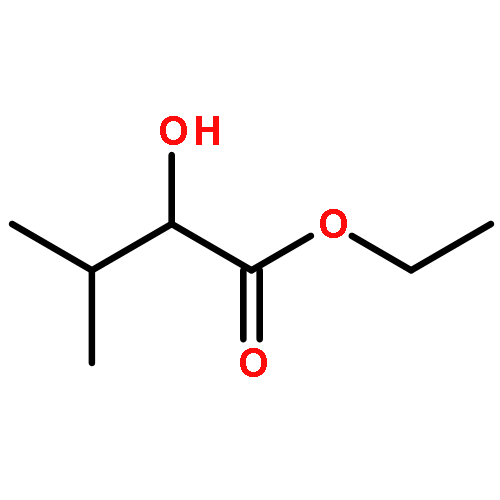
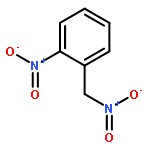

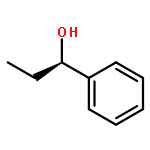
![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0.png)
![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0_b.png)

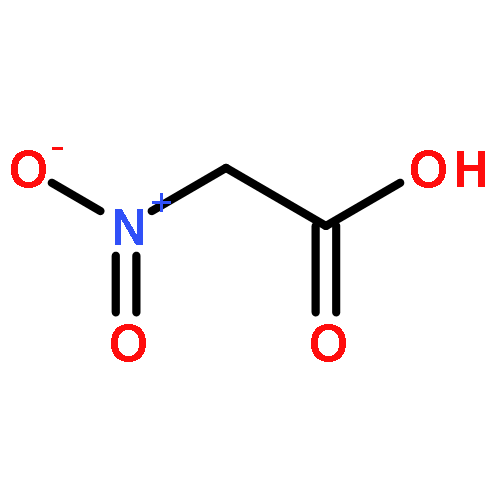
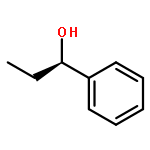
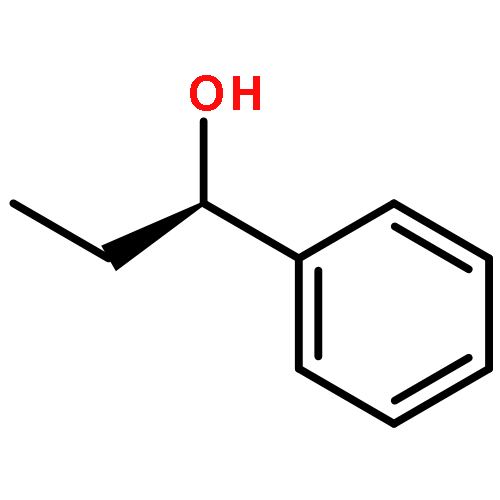
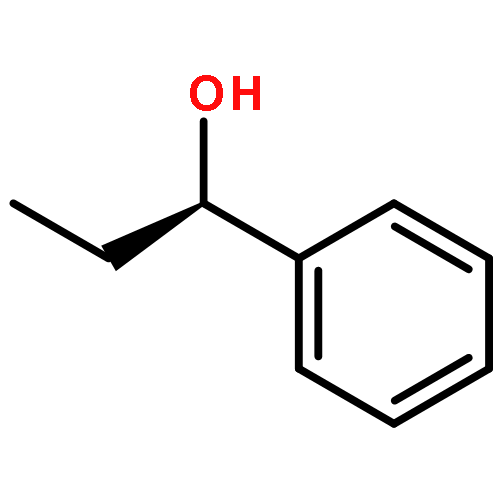
![Propanedioic acid, [(1R)-3-oxocyclohexyl]-, bis(phenylmethyl) ester](http://img.cochemist.com/ccimg/165000/164931-75-3.png)
![Propanedioic acid, [(1R)-3-oxocyclohexyl]-, bis(phenylmethyl) ester](http://img.cochemist.com/ccimg/165000/164931-75-3_b.png)
![BENZAMIDE, 2-[(1R)-1-METHYL-3-BUTENYL]-N,N-BIS(1-METHYLETHYL)-](http://img.cochemist.com/ccimg/159900/159888-32-1.png)
![BENZAMIDE, 2-[(1R)-1-METHYL-3-BUTENYL]-N,N-BIS(1-METHYLETHYL)-](http://img.cochemist.com/ccimg/159900/159888-32-1_b.png)




![Dichloro[(R)-(+)-2,2'-bis(diphenylphosphino)-1,1'-binaphthyl]palladium(II), min.](http://img.cochemist.com/ccimg/115900/115826-95-4.png)
![Dichloro[(R)-(+)-2,2'-bis(diphenylphosphino)-1,1'-binaphthyl]palladium(II), min.](http://img.cochemist.com/ccimg/115900/115826-95-4_b.png)




![5-HYDROXY-8,10-DIMETHOXY-2-METHYLBENZO[H]CHROMEN-4-ONE](http://img.cochemist.com/ccimg/3600/3566-99-2.png)
![5-HYDROXY-8,10-DIMETHOXY-2-METHYLBENZO[H]CHROMEN-4-ONE](http://img.cochemist.com/ccimg/3600/3566-99-2_b.png)


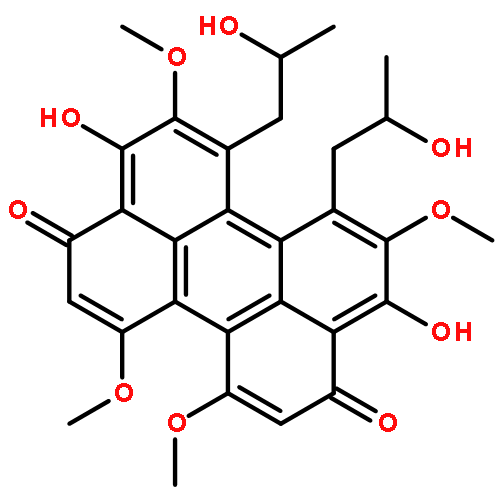
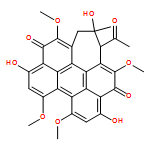
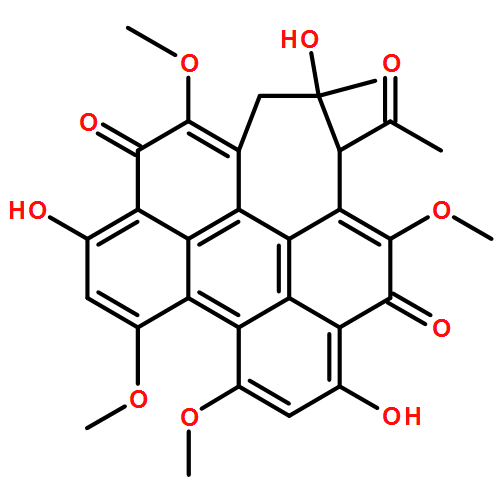
![1H-Cyclohepta[ghi]perylene-5,12-dione,1-acetyl-2,3-dihydro-2,6,11-trihydroxy-4,8,9,13-tetramethoxy-2-methyl-,stereoisomer](http://img.cochemist.com/ccimg/124800/124709-39-3.png)
![1H-Cyclohepta[ghi]perylene-5,12-dione,1-acetyl-2,3-dihydro-2,6,11-trihydroxy-4,8,9,13-tetramethoxy-2-methyl-,stereoisomer](http://img.cochemist.com/ccimg/124800/124709-39-3_b.png)
![5,5'-dihydroxy-6,6',8,8'-tetramethoxy-2,2'-dimethyl-4H,4'H-10,10'-bibenzo[g]chromene-4,4'-dione](http://img.cochemist.com/ccimg/76100/76069-41-5.png)
![5,5'-dihydroxy-6,6',8,8'-tetramethoxy-2,2'-dimethyl-4H,4'H-10,10'-bibenzo[g]chromene-4,4'-dione](http://img.cochemist.com/ccimg/76100/76069-41-5_b.png)


![Perylo[1,12-def]-1,3-dioxepin-5,11-dione,6,12-dihydroxy-8,9-bis[(2R)-2-hydroxypropyl]-7,10-dimethoxy-, (13bR)-](http://img.cochemist.com/ccimg/35100/35082-49-6.png)
![Perylo[1,12-def]-1,3-dioxepin-5,11-dione,6,12-dihydroxy-8,9-bis[(2R)-2-hydroxypropyl]-7,10-dimethoxy-, (13bR)-](http://img.cochemist.com/ccimg/35100/35082-49-6_b.png)
![3,10-Perylenedione,4,9-dihydroxy-1,12-bis[(2R)-2-hydroxypropyl]-2,6,7,11-tetramethoxy-, (6aS)-](http://img.cochemist.com/ccimg/125000/124986-26-1.png)
![3,10-Perylenedione,4,9-dihydroxy-1,12-bis[(2R)-2-hydroxypropyl]-2,6,7,11-tetramethoxy-, (6aS)-](http://img.cochemist.com/ccimg/125000/124986-26-1_b.png)




![[1,1'-Binaphthalene]-3,3'-dicarboxylicacid, 2,2'-dihydroxy-, 3,3'-dimethyl ester, (1R)-](http://img.cochemist.com/ccimg/18600/18531-91-4.png)
![[1,1'-Binaphthalene]-3,3'-dicarboxylicacid, 2,2'-dihydroxy-, 3,3'-dimethyl ester, (1R)-](http://img.cochemist.com/ccimg/18600/18531-91-4_b.png)
![Spiro[benzo[1,2-b:5,4-c']dipyran-2(3H),2'(3'H)-naphtho[2,3-b]furan]-7-carboxylicacid, 4,5',8',9-tetrahydro-4,4',9',10-tetrahydroxy-7'-methoxy-5',8',9-trioxo-,methyl ester, (2R,4S)-rel-](http://img.cochemist.com/ccimg/54000/53969-01-0.png)
![Spiro[benzo[1,2-b:5,4-c']dipyran-2(3H),2'(3'H)-naphtho[2,3-b]furan]-7-carboxylicacid, 4,5',8',9-tetrahydro-4,4',9',10-tetrahydroxy-7'-methoxy-5',8',9-trioxo-,methyl ester, (2R,4S)-rel-](http://img.cochemist.com/ccimg/54000/53969-01-0_b.png)