Co-reporter:Tianying Liu, Tyler M. Marcinko, Patrick A. Kiefer, and Richard W. Vachet
Analytical Chemistry November 7, 2017 Volume 89(Issue 21) pp:11583-11583
Publication Date(Web):October 13, 2017
DOI:10.1021/acs.analchem.7b02915
Amyloid aggregates are associated with several debilitating diseases, and there are numerous efforts to develop small molecule treatments against these diseases. One challenge associated with these efforts is determining protein binding site information for potential therapeutics because amyloid-forming proteins rapidly form oligomers and aggregates, making traditional protein structural analysis techniques challenging. Using β-2-microglobulin (β2m) as a model amyloid-forming protein along with two recently identified small molecule amyloid inhibitors (i.e., rifamycin SV and doxycycline), we demonstrate that covalent labeling and mass spectrometry (MS) can be used to map small-molecule binding sites for a rapidly aggregating protein. Specifically, three different covalent labeling reagents, namely diethylpyrocarbonate, 2,3-butanedione, and the reagent pair EDC/GEE, are used together to pinpoint the binding sites of rifamycin SV, doxycycline, and another molecule, suramin, which binds but does not inhibit Cu(II)-induced β2m amyloid formation. The labeling results reveal binding sites that are consistent with the known effects of these molecules on β2m amyloid formation and are in general agreement with molecular docking results. We expect that this combined covalent labeling approach will be applicable to other protein/small molecule systems that are difficult to study by traditional means.
Co-reporter:S. Gokhan Elci, Gulen Yesilbag Tonga, Bo Yan, Sung Tae Kim, Chang Soo Kim, Ying Jiang, Krishnendu Saha, Daniel F. Moyano, Alyssa L. M. Marsico, Vincent M. Rotello, and Richard W. Vachet
ACS Nano July 25, 2017 Volume 11(Issue 7) pp:7424-7424
Publication Date(Web):July 11, 2017
DOI:10.1021/acsnano.7b03711
Effective correlation of the in vitro and in vivo stability of nanoparticle-based platforms is a key challenge in their translation into the clinic. Here, we describe a dual imaging method that site-specifically reports the stability of monolayer-functionalized nanoparticles in vivo. This approach uses laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) imaging to monitor the distributions of the nanoparticle core material and laser desorption/ionization mass spectrometry (LDI-MS) imaging to report on the monolayers on the nanoparticles. Quantitative comparison of the images reveals nanoparticle stability at the organ and suborgan level. The stability of particles observed in the spleen was location-dependent and qualitatively similar to in vitro studies. In contrast, in vivo stability of the nanoparticles in the liver differed dramatically from in vitro studies, demonstrating the importance of in vivo assessment of nanoparticle stability.Keywords: dual imaging; in vivo monolayer stability; laser ablation inductively coupled plasma mass spectrometry; laser desorption/ionization mass spectrometry; nanoparticles;
Co-reporter:Zhe Zhang, Richard W. Vachet
International Journal of Mass Spectrometry 2017 Volume 420(Volume 420) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.ijms.2016.09.010
The gas phase structures of several proteins have been studied by electron transfer dissociation (ETD) with and without prior collisional heating after electrospraying these proteins from native-like solutions into a quadrupole ion trap mass spectrometer. Without prior collisional heating, we find that ETD fragmentation is mostly limited to regions of the protein that are not spanned by the salt bridges known to form in solution. When protein ions are collisionally heated before ETD, new product ions are observed, and in almost all cases, these new ions arise from protein regions that are spanned by the salt bridges. Together these results confirm the existence of salt bridges in protein ions and demonstrate that a sufficient amount energy is required to disrupt these salt bridges in the gas phase. More interestingly, we also show that different salt bridges require different collisional activation voltages to be disrupted, suggesting that they have variable stabilities in the gas phase. These stabilities appear to be influenced by the gas-phase basicities of the involved residues and the presence of nearby charged residues. We also find that higher collisional activation voltages are needed to enable the formation of new product from sites spanned by multiple salt bridges.Download high-res image (102KB)Download full-size image
Co-reporter:Mahalia A. C. Serrano;Huan He;Bo Zhao;Rajasekhar R. Ramireddy;S. Thayumanavan
Analyst (1876-Present) 2017 vol. 142(Issue 1) pp:118-122
Publication Date(Web):2016/12/19
DOI:10.1039/C6AN01591C
A combination of donor–acceptor and electrostatic interactions in a three-component supramolecular system has been shown to form the basis for selective and sensitive detection of peptides. Different substituents in the polymer and the detection matrix were compared to demonstrate that the favorable donor–acceptor interactions explain the observed signal enhancement. The ternary supramolecular interactions discovered in this work are enabled by the self-packing behavior of amphiphilic homopolymers and their ability to mediate interactions between the detection matrix and peptide that facilitate sensitive detection of peptides.
Co-reporter:Nicholas B. Borotto, Zhe Zhang, Jia Dong, Brittney Burant, and Richard W. Vachet
Biochemistry 2017 Volume 56(Issue 8) pp:
Publication Date(Web):February 7, 2017
DOI:10.1021/acs.biochem.6b01198
β-2-Microglobulin (β2m) forms amyloid fibrils in the joints of patients undergoing dialysis treatment as a result of kidney failure. One of the ways in which β2m can be induced to form amyloid fibrils in vitro is via incubation with stoichiometric amounts of Cu(II). To better understand the structural changes caused by Cu(II) binding that allow β2m to form amyloid fibrils, we compared the effect of Ni(II) and Zn(II) binding, which are two similarly sized divalent metal ions that do not induce β2m amyloid formation. Using hydrogen/deuterium exchange mass spectrometry (HDX/MS) and covalent labeling MS, we find that Ni(II) has little effect on β2m structure, despite binding in the same region of the protein as Cu(II). This observation indicates that subtle differences in the organization of residues around Cu(II) cause distant changes that are necessary for oligomerization and eventual amyloid formation. One key difference that we find is that only Cu(II), not Ni(II) or Zn(II), is able to cause the cis–trans isomerization of Pro32 that is an important conformational switch that initiates β2m amyloid formation. By comparing HDX/MS data from the three metal-β2m complexes, we also discover that increased dynamics in the β-sheet formed by the A, B, D, and E β strands of the protein and repositioning of residues in the D–E loop are necessary aspects of β2m forming an amyloid-competent dimer. Altogether, our results reveal new structural insights into the unique effect of Cu(II) in the metal-induced amyloid formation of β2m.
Co-reporter:Alyssa L. M. Marsico, Bradley Duncan, Ryan F. Landis, Gulen Yesilbag Tonga, Vincent M. RotelloRichard W. Vachet
Analytical Chemistry 2017 Volume 89(Issue 5) pp:
Publication Date(Web):February 2, 2017
DOI:10.1021/acs.analchem.6b04538
Nanomaterials have been extensively used as alternate matrices to minimize the low molecular weight interferences observed in typical MALDI but such nanomaterials typically do not improve the spot-to-spot variability that is commonly seen. In this work, we demonstrate that nanoparticles and low matrix concentrations (<2.5 mg/mL) can be used to homogeneously concentrate analytes into a narrow ring by taking advantage of the “coffee ring” effect. Concentration of the samples in this way leads to enhanced signals when compared to conventional MALDI, with higher m/z analytes being enhanced to the greatest extent. Moreover, the ionization suppression often observed in samples with high salt concentrations can be overcome by preparing samples in this way. The ring that is formed is readily visible, allowing the laser to be focused only on spots that contain analyte. The coffee-ring effect represents a new mode by which nanomaterials can be used to enhance the MALDI-based detection of biomolecules.
Co-reporter:S. Gokhan Elci, Bo Yan, Sung Tae Kim, Krishnendu Saha, Ying Jiang, Gunnar A. Klemmer, Daniel F. Moyano, Gulen Yesilbag Tonga, Vincent M. Rotello and Richard W. Vachet
Analyst 2016 vol. 141(Issue 8) pp:2418-2425
Publication Date(Web):10 Mar 2016
DOI:10.1039/C6AN00123H
Functionalized gold nanoparticles (AuNPs) have unique properties that make them important biomedical materials. Optimal use of these materials, though, requires an understanding of their fate in vivo. Here we describe the use of laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) to image the biodistributions of AuNPs in tissues from mice intravenously injected with AuNPs. We demonstrate for the first time that the distributions of very small (∼2 nm core) monolayer-protected AuNPs can be imaged in animal tissues at concentrations in the low parts-per-billion range. Moreover, the LA-ICP-MS images reveal that the monolayer coatings on the injected AuNPs influence their distributions, suggesting that the AuNPs remain intact in vivo and their surface chemistry influences how they interact with different organs. We also demonstrate that quantitative images of the AuNPs can be generated when the appropriate tissue homogenates are chosen for matrix matching. Overall, these results demonstrate the utility of LA-ICP-MS for tracking the fate of biomedically-relevant AuNPs in vivo, facilitating the design of improved AuNP-based therapeutics.
Co-reporter:Singyuk Hou, Kristen N. Sikora, Rui Tang, Yuanchang Liu, Yi-Wei Lee, Sung Tae Kim, Ziwen Jiang, Richard W. Vachet, and Vincent M. Rotello
ACS Nano 2016 Volume 10(Issue 7) pp:6731
Publication Date(Web):June 23, 2016
DOI:10.1021/acsnano.6b02105
Differentiation between cell surface-bound and internalized nanoparticles is challenging yet essential for accurately quantifying cellular uptake. Here, we describe a versatile mass spectrometry-based method that allows separate quantification of both cell surface-bound and internalized nanoparticles. This rapid method uses tuned laser fluencies to selectively desorb and ionize cell surface-bound cationic gold nanoparticles from intact cells, providing quantification of external particles. Overall nanoparticle quantities are obtained from the cell lysates, with subtraction of external particles from the total amount providing quantification of taken-up nanoparticles. The utility of this strategy was demonstrated through simultaneous quantitative determination of how cell-surface proteoglycans influence nanoparticle binding and uptake into cells.Keywords: cellular delivery; gold; mass spectrometry; nanoparticle
Co-reporter:Sukru Gokhan Elci, Ying Jiang, Bo Yan, Sung Tae Kim, Krishnendu Saha, Daniel F. Moyano, Gulen Yesilbag Tonga, Liam C. Jackson, Vincent M. Rotello, and Richard W. Vachet
ACS Nano 2016 Volume 10(Issue 5) pp:5536
Publication Date(Web):May 10, 2016
DOI:10.1021/acsnano.6b02086
Surface chemistry plays a deciding role in nanoparticle biodistribution, yet very little is known about how surface chemistry influences the suborgan distributions of nanomaterials. Here, using quantitative imaging based on laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS), we demonstrate that surface charge dictates the suborgan distributions of nanoparticles in the kidney, liver, and spleen of mice intravenously injected with functionalized gold nanoparticles. Images of the kidney show that positively charged nanoparticles accumulate extensively in the glomeruli, the initial stage in filtering for the nephron, suggesting that these nanoparticles may be filtered by the kidney at a different rate than the neutral or negatively charged nanoparticles. We find that positively and negatively charged nanoparticles accumulate extensively in the red pulp of the spleen. In contrast, uncharged nanoparticles accumulate in the white pulp and marginal zone of the spleen to a greater extent than the positively or negatively charged nanoparticles. Moreover, these uncharged nanoparticles are also more likely to be found associated with Kupffer cells in the liver. Positively charged nanoparticles accumulate extensively in liver hepatocytes, whereas negatively charged nanoparticles show a broader distribution in the liver. Together these observations suggest that neutral nanoparticles having 2 nm cores may interact with the immune system to a greater extent than charged nanoparticles, highlighting the value of determining the suborgan distributions of nanomaterials for delivery and imaging applications.Keywords: drug delivery; gold nanoparticle; imaging; LA-ICP-MS; suborgan biodistribution; surface functionality
Co-reporter:Nicholas B. Borotto, Yuping Zhou, Stephen R. Hollingsworth, John E. Hale, Eric M. Graban, Robert C. Vaughan, and Richard W. Vachet
Analytical Chemistry 2015 Volume 87(Issue 20) pp:10627
Publication Date(Web):September 23, 2015
DOI:10.1021/acs.analchem.5b03180
Protein therapeutics are rapidly transforming the pharmaceutical industry. Unlike for small molecule therapeutics, current technologies are challenged to provide the rapid, high-resolution analyses of protein higher order structures needed to ensure drug efficacy and safety. Consequently, significant attention has turned to developing new methods that can quickly, accurately, and reproducibly characterize the three-dimensional structure of protein therapeutics. In this work, we describe a method that uses diethylpyrocarbonate (DEPC) labeling and mass spectrometry to detect three-dimensional structural changes in therapeutic proteins that have been exposed to degrading conditions. Using β2-microglobulin, immunoglobulin G1, and human growth hormone as model systems, we demonstrate that DEPC labeling can identify both specific protein regions that mediate aggregation and those regions that undergo more subtle structural changes upon mishandling of these proteins. Importantly, DEPC labeling is able to provide information for up to 30% of the surface residues in a given protein, thereby providing excellent structural resolution. Given the simplicity of the DEPC labeling chemistry and the relatively straightforward mass spectral analysis of DEPC-labeled proteins, we expect this method should be amenable to a wide range of protein therapeutics and their different formulations.
Co-reporter:Zhe Zhang and Richard W. Vachet
Analytical Chemistry 2015 Volume 87(Issue 23) pp:11777
Publication Date(Web):November 4, 2015
DOI:10.1021/acs.analchem.5b03123
The growing importance of protein aggregation diseases requires the development of new methods to elucidate the molecular features that are responsible for the incipient protein–protein interactions. Kinetic information from protein–protein association/dissociation reactions is particularly valuable for revealing mechanistic insight, but robust tools that can provide this information are somewhat lacking. In this work, we describe a hydrogen/deuterium exchange (HDX)-based method that provides rate constant information for protein oligomer dissociation, using the well-studied β-lactoglobulin (βLG) dimer as a model system to validate our approach. By measuring the rate of exchange at different regions of the protein using top-down tandem mass spectrometry and fitting the resulting data to an appropriate mathematical model, we are able to extract the dimer’s dissociation rate constant. We exploit the fact that regions of the protein that are part of the protein–protein interface have exchange patterns that are distinct from noninterfacial regions. This observation indicates that the HDX/MS method not only provides kinetic information but also could provide structural insight about the interface at the same time, which would be very valuable for previously uncharacterized protein–protein complexes.
Co-reporter:Alyssa L. M. Marsico, Gokhan S. Elci, Daniel F. Moyano, Gulen Yesilbag Tonga, Bradley Duncan, Ryan F. Landis, Vincent M. Rotello, and Richard W. Vachet
Analytical Chemistry 2015 Volume 87(Issue 24) pp:12145
Publication Date(Web):November 11, 2015
DOI:10.1021/acs.analchem.5b02985
Laser desorption/ionization mass spectrometry (LDI-MS) has been used to detect gold nanoparticles (AuNPs) in biological samples, such as cells and tissues, by ionizing their attached monolayer ligands. Many NP-attached ligands, however, are difficult to ionize by LDI, making it impossible to track these NPs in biological samples. In this work, we demonstrate that concentrations of matrix-assisted LDI (MALDI) matrices an order of magnitude below the values typically used in MALDI can facilitate the selective detection of AuNPs with these ligands, even in samples as complex as cell lysate. This enhanced sensitivity arises from a synergistic relationship between the gold core and the matrix that helps to selectively ionize ligands attached to the AuNPs.
Co-reporter:Alyssa L. M. Marsico;Brian Creran
Journal of The American Society for Mass Spectrometry 2015 Volume 26( Issue 11) pp:1931-1937
Publication Date(Web):2015 November
DOI:10.1007/s13361-015-1223-x
Effective detection of low molecular weight compounds in matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS) is often hindered by matrix interferences in the low m/z region of the mass spectrum. Here, we show that monolayer-protected gold nanoparticles (AuNPs) can serve as alternate matrices for the very sensitive detection of low molecular weight compounds such as amino acids. Amino acids can be detected at low fmol levels with minimal interferences by properly choosing the AuNP deposition method, density, size, and monolayer surface chemistry. By inkjet-printing AuNPs at various densities, we find that AuNP clusters are essential for obtaining the greatest sensitivity.
Co-reporter:Bo Yan, Gulen Yesilbag Tonga, Singyuk Hou, Patrick W. Fedick, Yi-Cheun Yeh, Felix S. Alfonso, Tsukasa Mizuhara, Richard W. Vachet, and Vincent M. Rotello
Analytical Chemistry 2014 Volume 86(Issue 13) pp:6710
Publication Date(Web):May 29, 2014
DOI:10.1021/ac501682y
Synthetic host–guest chemistry is a versatile tool for biomedical applications. Characterization and detection of host–guest complexes in biological systems, however, is challenging due to the complexity of the biological milieu. Here, we describe and apply a mass spectrometric method to monitor the association and dissociation of nanoparticle (NP)-based host–guest interactions that integrates NP-assisted laser desorption/ionization (LDI) and matrix assisted laser desoption/ionization (MALDI) mass spectrometry. This LDI/MALDI approach reveals how NP surface functionality affects host–guest interactions in cells, information difficult to achieve using other techniques.
Co-reporter:Jia Dong, Katie L. Callahan, Nicholas B. Borotto, and Richard W. Vachet
Analytical Chemistry 2014 Volume 86(Issue 1) pp:766
Publication Date(Web):December 7, 2013
DOI:10.1021/ac4032719
In this work, we have developed a method that uses hydrogen–deuterium exchange (HDX) of C2-hydrogens of histidines coupled with mass spectrometry (MS) to identify Zn-bound histidines in metalloproteins. This method relies on differences in HDX reaction rates of Zn-bound and Zn-free His residues. Using several model peptides and proteins, we find that all Zn-bound His residues have substantially lower HDX reaction rates in the presence of the metal. The vast majority of non-Zn-binding His residues undergo no significant changes in HDX reaction rates when their reactivity is compared in the presence and absence of Zn. Using this new approach, we then determined the Zn binding site of β-2-microglobulin, a protein associated with metal-induced amyloidosis. Together, these results suggest that HDX-MS of His C2-hydrogens is a promising new method for identifying Zn-bound histidines in metalloproteins.
Co-reporter:Jia Dong, Crisjoe A. Joseph, Nicholas B. Borotto, Vanessa L. Gill, Michael J. Maroney, and Richard W. Vachet
Biochemistry 2014 Volume 53(Issue 8) pp:
Publication Date(Web):January 22, 2014
DOI:10.1021/bi4016583
β-2-Microglobulin (β2m) forms amyloid fibrils in the joints of patients undergoing hemodialysis treatment as a result of kidney failure. In the presence of stoichiometric amounts of Cu(II), β2m self-associates into discrete oligomeric species, including dimers, tetramers, and hexamers, before ultimately forming amyloid fibrils that contain no copper. To improve our understanding of whether Cu(II) is unique in its ability to induce β2m amyloid formation and to delineate the coordinative interactions that allow Cu(II) to exert its effect, we have examined the binding of Ni(II) and Zn(II) to β2m and the resulting influence that these metals have on β2m aggregation. We find that, in contrast to Cu(II), Ni(II) does not induce the oligomerization or aggregation of β2m, while Zn(II) promotes oligomerization but not amyloid fibril formation. Using X-ray absorption spectroscopy and new mass spectrometry-related techniques, we find that different binding modes are responsible for the different effects of Ni(II) and Zn(II). By comparing the binding modes of Cu(II) with Ni(II), we find that Cu(II) binding to Asp59 and the backbone amide between the first two residues of β2m are important for allowing the formation of amyloid-competent oligomers, as Ni(II) appears not to bind these sites on the protein. The oligomers formed in the presence of Zn(II) are permitted by this metal’s ability to bridge two β2m units via His51. These oligomers, however, are not able to progress to form amyloid fibrils because Zn(II) does not induce the required structural changes near the N-terminus and His31.
Co-reporter:Zhe Zhang;Shaynah J. Browne
Journal of The American Society for Mass Spectrometry 2014 Volume 25( Issue 4) pp:604-613
Publication Date(Web):2014 April
DOI:10.1007/s13361-013-0821-8
The gas-phase structures of protein ions have been studied by electron transfer dissociation (ETD) and collision-induced dissociation (CID) after electrospraying these proteins from native-like solutions into a quadrupole ion trap mass spectrometer. Because ETD can break covalent bonds while minimally disrupting noncovalent interactions, we have investigated the ability of this dissociation technique together with CID to probe the sites of electrostatic interactions in gas-phase protein ions. By comparing spectra from ETD with spectra from ETD followed by CID, we find that several proteins, including ubiquitin, CRABP I, azurin, and β-2-microglobulin, appear to maintain many of the salt bridge contacts known to exist in solution. To support this conclusion, we also performed calculations to consider all possible salt bridge patterns for each protein, and we find that the native salt bridge pattern explains the experimental ETD data better than nearly all other possible salt bridge patterns. Overall, our data suggest that ETD and ETD/CID of native protein ions can provide some insight into approximate location of salt bridges in the gas phase.
Co-reporter:Nicholas B. Borotto;Nicholas Degraan-Weber
Journal of The American Society for Mass Spectrometry 2014 Volume 25( Issue 10) pp:1739-1746
Publication Date(Web):2014 October
DOI:10.1007/s13361-014-0962-4
Covalent labeling along with mass spectrometry is finding more use as a means of studying the higher order structure of proteins and protein complexes. Diethylpyrocarbonate (DEPC) is an increasingly used reagent for these labeling experiments because it is capable of modifying multiple residues at the same time. Pinpointing DEPC-labeled sites on proteins is typically needed to obtain more resolved structural information, and tandem mass spectrometry after protein proteolysis is often used for this purpose. In this work, we demonstrate that in certain instances, scrambling of the DEPC label from one residue to another can occur during collision-induced dissociation (CID) of labeled peptide ions, resulting in ambiguity in label site identity. From a preliminary study of over 30 labeled peptides, we find that scrambling occurs in about 25% of the peptides and most commonly occurs when histidine residues are labeled. Moreover, this scrambling appears to occur more readily under non-mobile proton conditions, meaning that low charge-state peptide ions are more prone to this reaction. For all peptides, we find that scrambling does not occur during electron transfer dissociation, which suggests that this dissociation technique is a safe alternative to CID for correct label site identification.
Co-reporter:Bo Yan ; Sung Tae Kim ; Chang Soo Kim ; Krishnendu Saha ; Daniel F. Moyano ; Yuqing Xing ; Ying Jiang ; Amy L. Roberts ; Felix S. Alfonso ; Vincent M. Rotello
Journal of the American Chemical Society 2013 Volume 135(Issue 34) pp:12564-12567
Publication Date(Web):August 9, 2013
DOI:10.1021/ja406553f
Imaging of nanomaterials in biological tissues provides vital information for the development of nanotherapeutics and diagnostics. Multiplexed imaging of different nanoparticles (NPs) greatly reduces costs, the need to use multiple animals, and increases the biodistribution information that can enhance diagnostic applications and accelerate the screening of potential therapeutics. Various approaches have been developed for imaging NPs; however, the readout of existing imaging techniques relies on specific properties of the core material or surface ligands, and these techniques are limited because of the relatively small number of NPs that can be simultaneously measured in a single experiment. Here, we demonstrate the use of laser desorption/ionization mass spectrometry (LDI-MS) in an imaging format to investigate surface chemistry dictated intraorgan distribution of NPs. This new LDI-MS imaging method enables multiplexed imaging of NPs with potentially unlimited readouts and without additional labeling of the NPs. It provides the capability to detect and image attomole levels of NPs with almost no interferences from biomolecules. Using this new imaging approach, we find that the intraorgan distributions of same-sized NPs are directly linked to their surface chemistry.
Co-reporter:Feng Wang ; Andrea Gomez-Escudero ; Rajasekhar R. Ramireddy ; Gladys Murage ; S. Thayumanavan
Journal of the American Chemical Society 2013 Volume 135(Issue 38) pp:14179-14188
Publication Date(Web):August 23, 2013
DOI:10.1021/ja404940s
Supramolecular assemblies formed by amphiphilic homopolymers with negatively charged groups in the hydrophilic segment have been designed to enable high labeling selectivity toward reactive side chain functional groups in peptides. The negatively charged interiors of the supramolecular assemblies are found to block the reactivity of protonated amines that would otherwise be reactive in aqueous solution, while maintaining the reactivity of nonprotonated amines. Simple changes to the pH of the assemblies’ interiors allow control over the reactivity of different functional groups in a manner that is dependent on the pKa of a given peptide functional group. The labeling studies carried out in positively charged supramolecular assemblies and free buffer solution show that, even when the amine is protonated, labeling selectivity exists only when complementary electrostatic interactions are present, thereby demonstrating the electrostatically controlled nature of these reactions.
Co-reporter:Bo Yan, Youngdo Jeong, Luiza A. Mercante, Gülen Yesilbag Tonga, Chaekyu Kim, Zheng-Jiang Zhu, Richard W. Vachet and Vincent M. Rotello
Nanoscale 2013 vol. 5(Issue 11) pp:5063-5066
Publication Date(Web):18 Apr 2013
DOI:10.1039/C3NR01384G
Functionalized magnetic nanoparticles (MNPs) have been characterized by laser desorption/ionization mass spectrometry (LDI-MS). Quantitative information about surface ligand composition and structure for monolayer and mixed monolayer protected Fe3O4 and FePt NPs can be obtained rapidly with very little sample consumption.
Co-reporter:Yuping Zhou and Richard W. Vachet
Analytical Chemistry 2013 Volume 85(Issue 20) pp:9664
Publication Date(Web):September 6, 2013
DOI:10.1021/ac401978w
Covalent labeling and mass spectrometry (MS) are increasingly being used to obtain higher-order structure of proteins and protein complexes. Because most covalent labels are relatively large, steps must be taken to ensure the structural integrity of the modified protein during the labeling reactions so that correct structural information can be obtained. Measuring labeling kinetics is a reliable way to ensure that a given labeling reagent does not perturb a protein’s structure, but obtaining such kinetic information is time and sample intensive because it requires multiple liquid chromatography (LC)–MS experiments. Here we present a new strategy that uses isotopically encoded labeling reagents to measure labeling kinetics in a single LC–MS experiment. We illustrate this new strategy by labeling solvent-exposed lysine residues with commercially available tandem mass tags. After tandem MS experiments, these tags allow the simultaneous identification of modified sites and determination of the reaction rates at each site in a way that is just as reliable as experiments that involve multiple LC–MS measurements.
Co-reporter:Adam M. Graichen
Journal of The American Society for Mass Spectrometry 2013 Volume 24( Issue 6) pp:917-925
Publication Date(Web):2013 June
DOI:10.1007/s13361-013-0592-2
The gas-phase reactions of a series of coordinatively unsaturated [Ni(L)n]y+ complexes, where L is a nitrogen-containing ligand, with chemical warfare agent (CWA) simulants in a miniature rectilinear ion trap mass spectrometer were investigated as part of a new approach to detect CWAs. Results show that upon entering the vacuum system via a poly(dimethylsiloxane) (PDMS) membrane introduction, low concentrations of several CWA simulants, including dipropyl sulfide (simulant for mustard gas), acetonitrile (simulant for the nerve agent tabun), and diethyl phosphite (simulant for nerve agents sarin, soman, tabun, and VX), can react with metal complex ions generated by electrospray ionization (ESI), thereby providing a sensitive means of detecting these compounds. The [Ni(L)n]2+ complexes are found to be particularly reactive with the simulants of mustard gas and tabun, allowing their detection at low parts-per-billion (ppb) levels. These detection limits are well below reported exposure limits for these CWAs, which indicates the applicability of this new approach, and are about two orders of magnitude lower than electron ionization detection limits on the same mass spectrometer. The use of coordinatively unsaturated metal complexes as reagent ions offers the possibility of further tuning the ion-molecule chemistry so that desired compounds can be detected selectively or at even lower concentrations.
Co-reporter:Zheng-Jiang Zhu, Rui Tang, Yi-Cheun Yeh, Oscar R. Miranda, Vincent M. Rotello, and Richard W. Vachet
Analytical Chemistry 2012 Volume 84(Issue 10) pp:4321
Publication Date(Web):April 20, 2012
DOI:10.1021/ac203408v
Monolayer stability of core–shell nanoparticles is a key determinant of their utility in biological studies such as imaging and drug delivery. Intracellular thiols (e.g., cysteine, cysteamine, and glutathione) can trigger the release of thiolate-bound monolayers from nanoparticles, a favorable outcome for controllable drug release applications but an unfavorable outcome for imaging agents. Here, we describe a method to quantify the monolayer release of gold nanoparticles (AuNPs) in living cells using parallel measurements by laser desorption/ionization (LDI) and inductively coupled plasma (ICP) mass spectrometry. This combination of methods is tested using AuNPs with structural features known to influence monolayer stability and on cells types with varying concentrations of glutathione. On the basis of our results, we predict that this approach should help efforts to engineer nanoparticle surface monolayers with tunable stability, providing stable platforms for imaging agents and controlled release of therapeutic monolayer payloads.
Co-reporter:Zheng-Jiang Zhu, Huanhua Wang, Bo Yan, Hao Zheng, Ying Jiang, Oscar R. Miranda, Vincent M. Rotello, Baoshan Xing, and Richard W. Vachet
Environmental Science & Technology 2012 Volume 46(Issue 22) pp:12391
Publication Date(Web):October 26, 2012
DOI:10.1021/es301977w
Small (6–10 nm) functionalized gold nanoparticles (AuNPs) featuring different, well-defined surface charges were used to probe the uptake and distribution of nanomaterials in terrestrial plants, including rice, radish, pumpkin, and perennial ryegrass. Exposure of the AuNPs to plant seedlings under hydroponic conditions for a 5-day period was investigated. Results from these studies indicate that AuNP uptake and distribution depend on both nanoparticle surface charge and plant species. The experiments show that positively charged AuNPs are most readily taken up by plant roots, while negatively charged AuNPs are most efficiently translocated into plant shoots (including stems and leaves) from the roots. Radish and ryegrass roots generally accumulated higher amounts of the AuNPs (14–900 ng/mg) than rice and pumpkin roots (7–59 ng/mg). Each of the AuNPs used in this study were found to accumulate to statistically significant extents in rice shoots (1.1–2.9 ng/mg), while none of the AuNPs accumulated in the shoots of radishes and pumpkins.
Co-reporter:Nadnudda Rodthongkum, Rajasekhar Ramireddy, S. Thayumanavan and W. Vachet Richard
Analyst 2012 vol. 137(Issue 4) pp:1024-1030
Publication Date(Web):23 Dec 2011
DOI:10.1039/C2AN16089G
Reverse-micelle forming amphiphilic homopolymers with carboxylic acid and quaternary amine substituents are used to selectively enrich biomarker peptides and protein fragments from human serum prior to matrix assisted laser desorption/ionization mass spectrometry (MALDI-MS) analysis. After depletion of human serum albumin (HSA) and immunoglobulin G (IgG), low abundance peptide biomarkers can be selectively enriched and detected by MALDI-MS at clinically relevant concentrations by using the appropriate homopolymer(s) and extraction pH value(s). Three breast cancer peptide biomarkers, bradykinin, C4a, and ITIH4, were chosen to test this new approach, and detection limits of 0.5 ng mL−1, 0.08 ng mL−1, and 0.2 ng mL−1, respectively, were obtained. In addition, the amphiphilic homopolymers were used to detect prostate specific antigen (PSA) at concentrations as low as 0.5 ng mL−1 by targeting a surrogate peptide fragment of this protein biomarker. Selective enrichment and sensitive MS detection of low abundance peptide/protein biomarkers by these polymeric reverse micelles should be a sensitive and straightforward approach for biomarker screening in human serum.
Co-reporter:Yuping Zhou
Journal of The American Society for Mass Spectrometry 2012 Volume 23( Issue 4) pp:708-717
Publication Date(Web):2012 April
DOI:10.1007/s13361-011-0332-4
Covalent labeling and mass spectrometry are seeing increased use together as a way to obtain insight into the 3-dimensional structure of proteins and protein complexes. Several amino acid specific (e.g., diethylpyrocarbonate) and non-specific (e.g., hydroxyl radicals) labeling reagents are available for this purpose. Diethylpyrocarbonate (DEPC) is a promising labeling reagent because it can potentially probe up to 30% of the residues in the average protein and gives only one reaction product, thereby facilitating mass spectrometric analysis. It was recently reported, though, that DEPC modifications are labile for some amino acids. Here, we show that label loss is more significant and widespread than previously thought, especially for Ser, Thr, Tyr, and His residues, when relatively long protein digestion times are used. Such label loss ultimately decreases the amount of protein structural information that is obtainable with this reagent. We find, however, that the number of DEPC modified residues and, thus, protein structural information, can be significantly increased by decreasing the time between the covalent labeling reaction and the mass spectrometric analysis. This is most effectively accomplished using short (e.g., 2 h) proteolytic digestions with enzymes such as immobilized chymotrypsin or Glu-C rather than using methods (e.g., microwave or ultrasonic irradiation) that accelerate proteolysis in other ways. Using short digestion times, we show that the percentage of solvent accessible residues that can be modified by DEPC increases from 44% to 67% for cytochrome c, 35% to 81% for myoglobin, and 76% to 95% for β-2-microglobulin. In effect, these increased numbers of modified residues improve the protein structural resolution available from this covalent labeling method. Compared with typical overnight digestion conditions, the short digestion times decrease the average distance between modified residues from 11 to 7 Å for myoglobin, 13 to 10 Å for cytochrome c, and 9 to 8 Å for β-2-microglobulin.
Co-reporter:Jia Dong
Journal of The American Society for Mass Spectrometry 2012 Volume 23( Issue 2) pp:321-329
Publication Date(Web):2012 February
DOI:10.1007/s13361-011-0299-1
In contrast to previous electron capture dissociation (ECD) studies, we find that electron transfer dissociation (ETD) of Cu(II)–peptide complexes can generate c- and z-type product ions when the peptide has a sufficient number of strongly coordinating residues. Double-resonance experiments, ion-molecule reactions, and collision-induced dissociation (CID) prove that the c and z product ions are formed via typical radical pathways without the associated reduction of Cu(II), despite the high second ionization energy of Cu. A positive correlation between the number of Cu(II) binding groups in the peptide sequence and the extent of c and z ion formation was also observed. This trend is rationalized by considering that the recombination energy of Cu(II) can be lowered by strong binding ligands to an extent that enables electron transfer to non-Cu sites (e.g., protonation sites) to compete with Cu(II) reduction, thereby generating c/z ions in a manner similar to that observed for protonated (i.e., nonmetalated) peptides.
Co-reporter:Yuping Zhou
Journal of The American Society for Mass Spectrometry 2012 Volume 23( Issue 5) pp:899-907
Publication Date(Web):2012 May
DOI:10.1007/s13361-012-0349-3
Covalent labeling along with mass spectrometry is a method that is increasingly used to study protein structure. Recently, it has been shown that diethylpyrocarbonate (DEPC) is a powerful labeling reagent because it can modify up to 30% of the residues in the average protein, including the N-terminus, His, Lys, Tyr, Ser, Thr, and Cys residues. We recently discovered, however, that Cys residues that form disulfide bonds appear to be modified by DEPC as well. In this work, we demonstrate that disulfide linked Cys residues are not actually reactive with DEPC but, instead, once reduced, free Cys residues can capture a carbethoxy group from other modified amino acids via a solution-phase reaction that can occur during the protein digestion step. This “scrambling” of carbethoxy groups decreases the amount of modification observed at other residues and can potentially provide incorrect protein structural information. Fortunately, label scrambling can be completely avoided by alkylating the free thiols after disulfide reduction.
Co-reporter:Vanessa Leah Mendoza, Mario A. Barón-Rodríguez, Cristian Blanco, and Richard W. Vachet
Biochemistry 2011 Volume 50(Issue 31) pp:
Publication Date(Web):June 30, 2011
DOI:10.1021/bi2004894
The main pathogenic process underlying dialysis-related amyloidosis is the accumulation of β-2-microglobulin (β2m) as amyloid fibrils in the musculoskeletal system, and some evidence suggests that Cu(II) may play a role in β2m amyloid formation. Cu(II)-induced β2m fibril formation is preceded by the formation of discrete, oligomeric intermediates, including dimers, tetramers, and hexamers. In this work, we use selective covalent labeling reactions combined with mass spectrometry to investigate the amino acids responsible for mediating tetramer formation in wild-type β2m. By comparing the labeling patterns of the monomer, dimer, and tetramer, we find evidence that the tetramer interface is formed by the interaction of D strands from one dimer unit and G strands from another dimer unit. These covalent labeling data along with molecular dynamics calculations allow the construction of a tetramer model that indicates how the protein might proceed to form even higher-order oligomers.
Co-reporter:Adam M. Graichen
Journal of The American Society for Mass Spectrometry 2011 Volume 22( Issue 4) pp:683-688
Publication Date(Web):2011 April
DOI:10.1007/s13361-010-0068-6
A multiplexed method for performing MS/MS on multiple ions simultaneously in a miniature rectilinear ion trap (RIT) mass spectrometer has been developed. This method uses an ion encoding procedure that relies on the mass bias that exists when ions are externally injected into an RIT operated with only a single phase rf applied to one pair of electrodes. The ion injection profile under such conditions ions is Gaussian-like over a wide range of rf amplitudes, or low mass cutoff (LMCO) values, during ion accumulation. We show that this distribution is related to ion m/z and is likely caused by ions having an optimal range of pseudo-potential well depths for efficient trapping. Based on this observation, precursor ion intensity changes between two different injection LMCO values can be predicted, and these ion intensity changes are found to be carried through to their corresponding product ions, enabling multiplexed MS/MS spectra to be deconvoluted.
Co-reporter:Nadnudda Rodthongkum, Yangbin Chen, S. Thayumanavan, and Richard W. Vachet
Analytical Chemistry 2010 Volume 82(Issue 20) pp:8686
Publication Date(Web):September 23, 2010
DOI:10.1021/ac101922b
The typical difficulties associated with the detection of acidic peptides (i.e., those with low isoelectric points (pI)) by matrix-assisted laser desorption/ionization-mass spectrometry (MALDI-MS) represent a challenge in some proteomic analyses. Here, reverse micelle-forming amphiphilic homopolymers with positively charged interiors are synthesized and used to selectively enrich low pI peptides from complex mixtures for MALDI-MS detection. When using these polymers, acidic proteolytic peptides that are undetectable during normal MALDI-MS analysis are selectively detected. We show that enrichment of these low pI peptides allows acidic proteins to be selectively targeted for detection in multiprotein digests. In addition, the presence of the positively charged polymers during MALDI-MS analyses enhances peptide ion signals by almost an order of magnitude, thereby achieving reproducible ion signals for acidic peptides at concentrations as low as 100 fM. Concurrent detection of acidic and basic peptides was also facilitated by utilizing a sequential extraction process involving reverse micelle forming polymers with positively and negatively charged interiors.
Co-reporter:Nadnudda Rodthongkum, Yangbin Chen, S. Thayumanavan and Richard W. Vachet
Analytical Chemistry 2010 Volume 82(Issue 9) pp:3686
Publication Date(Web):April 8, 2010
DOI:10.1021/ac1000256
Extraction of peptides by reverse micelle-forming amphiphilic homopolymers and subsequent matrix-assisted laser desorption ionization-mass spectrometry (MALDI-MS) detection of these peptides in the presence of these polymers can significantly enhance peptide ion signals. Here, the mechanism of this MALDI signal enhancement is investigated. We find that the signal enhancement is caused by coalescence of polymer−peptide conjugates into “hotspots” on the MALDI target. Hotspot formation is observed only on hydrophilic surfaces and not hydrophobic surfaces. With the use of an Anchorchip MALDI target, which contains very small hydrophilic spots surrounded by a larger hydrophobic area, we find that this hotspot formation can be further exploited for ultrasensitive MALDI-MS analyses of peptides and peptide mixtures. MALDI-MS signals can be enhanced by 3−5 orders of magnitude when peptides are extracted by the amphiphilic homopolymers and detected on the Anchorchip MALDI target. This signal enhancement combined with the extraction selectivity of these reverse micelle-forming homopolymers makes these materials promising tools for sensitive detection of peptides in complex mixtures.
Co-reporter:Vanessa Leah Mendoza, Kwasi Antwi, Mario A. Barón-Rodríguez, Cristian Blanco and Richard W. Vachet
Biochemistry 2010 Volume 49(Issue 7) pp:
Publication Date(Web):January 20, 2010
DOI:10.1021/bi901748h
β-2-Microglobulin (β2m) self-associates into fibrillar amyloid deposits in the musculoskeletal system of patients undergoing hemodialysis treatment. Previous studies have shown that stoichiometric amounts of Cu(II) at near physiological conditions can cause β2m to organize into native-like dimers prior to forming amyloid fibrils. Here, we report the results from selective covalent labeling reactions combined with mass spectrometry that provide insight into the amino acid residues that mediate dimer formation in the wild-type protein. Using three complementary covalent labeling reagents, we find that the dimer interface is formed by the antiparallel stacking of ABED β-sheets from two β2m monomers. In addition, our data clearly indicate that a dimer interface involving the interactions of D−D strands from separate protein units as seen in the recent crystal structures of two mutant β2m oligomers is unlikely.
Co-reporter:Bo Yan;Zheng-Jiang Zhu;Oscar R. Miranda
Analytical and Bioanalytical Chemistry 2010 Volume 396( Issue 3) pp:1025-1035
Publication Date(Web):2010 February
DOI:10.1007/s00216-009-3250-6
Monolayer-protected gold nanoparticles (AuNPs) feature unique surface properties that enable numerous applications. Thus, there is a need for simple, rapid, and accurate methods to confirm the surface structures of these materials. Here, we describe how laser desorption/ionization mass spectrometry (LDI-MS) can be used to characterize AuNPs with neutral, positively, and negatively charged surface functional groups. LDI readily desorbs and ionizes the gold-bound ligands to produce both free thiols and disulfide ions in pure and complex samples. We also find that LDI-MS can provide a semi-quantitative measure of the ligand composition of mixed-monolayer AuNPs by monitoring mixed disulfide ions that are formed. Overall, the LDI-MS approach requires very little sample, provides an accurate measure of the surface ligands, and can be used to monitor AuNPs in complex mixtures.
Co-reporter:Nadnudda Rodthongkum, Jacqueline D. Washington, Elamprakash N. Savariar, S. Thayumanavan and Richard W. Vachet
Analytical Chemistry 2009 Volume 81(Issue 12) pp:5046
Publication Date(Web):May 21, 2009
DOI:10.1021/ac900661e
Amphiphilic homopolymers that self-assemble into reverse micelles in nonpolar solvents have been used by us in the context of a two-phase liquid−liquid extraction protocol to selectively extract peptides from aqueous solution for MALDI-MS detection. In this manuscript, we investigate the scope of these materials in terms of its extraction capabilities, using compounds with varying isoelectric points (pI) and pKa values over a range of aqueous solution pHs. We find that the aqueous solution pH and analyte pKa values are the major factors controlling extraction selectivity. We also find that the experimental extraction efficiencies correspond very well with the fractional compositions of species calculated using analyte pKa values, indicating that these extraction materials can be used to simultaneously generate titration-type curves for each individual peptide in a mixture. We predict that such titration curves, along with accurate mass measurements, could represent a new way of improving protein identification procedures.
Co-reporter:Zheng-Jiang Zhu, Vincent M. Rotello and Richard W. Vachet
Analyst 2009 vol. 134(Issue 11) pp:2183-2188
Publication Date(Web):21 Sep 2009
DOI:10.1039/B910428C
Engineering of nanoparticle surface functionality provides controlled interactions with biomolecules such as cell membrane lipids, proteins and nucleic acids. Concurrently, this surface chemistry control also opens up new avenues for improving mass spectral analyses. In this Minireview, we highlight some of the emerging work that integrates surface-engineered nanoparticles with mass spectrometry to improve the analysis of a wide variety of chemical and biological systems.
Co-reporter:Rapole Srikanth, Vanessa Leah Mendoza, Juma D. Bridgewater, Guanshi Zhang and Richard W. Vachet
Biochemistry 2009 Volume 48(Issue 41) pp:
Publication Date(Web):September 15, 2009
DOI:10.1021/bi901172y
β-2-Microglobulin (β2m) deposits as amyloid fibrils in the musculoskeletal system of patients undergoing long-term dialysis treatment as a result of kidney failure. Previous work has shown that Cu(II) binding causes β2m to organize into nativelike dimers and tetramers that precede amyloid formation. Cu(II) is then released from higher-order oligomers before mature Cu(II)-free amyloid fibrils are formed. While some of the Cu(II)-induced structural changes that enable β2m self-assembly are starting to be revealed, the details of how the Cu(II) binding site evolves from the monomer to the dimers and tetramers are not known. Here, we report results from three mass spectrometry (MS)-based methods that provide insight into the changing Cu−β2m interactions. We find that monomeric β2m binds Cu(II) via the N-terminal amine, the amide of Gln2, His31, and Asp59. In the dimer and tetramer, Asp59 is no longer bound to Cu(II), but the other residues still comprise a well-defined albeit weaker binding site that is better able to release Cu(II). Consistent with this is the observation that a fraction of the tetrameric species no longer binds Cu(II) at this weakened binding site, which agrees with a previous report that suggested the tetramer as the first Cu(II)-free oligomer. Our results also provide some insight into structural changes caused by Cu(II) binding that facilitate oligomer formation. Specifically, binding by Asp59 in the monomer requires significant movement of this residue, and we propose that this repositioning is important for establishing a pair of dimer-stabilizing salt bridges between this residue and Lys19. We also find evidence that Cu(II) binding in the N-terminal region of the monomer repels Arg3, which likely allows this residue to form a pair of dimer-stabilizing salt bridges with Glu16. Overall, our measurements suggest that the previously proposed conformational switch caused by Cu(II) binding includes not only a cis−trans isomerization at Pro32 but also the repositioning of residues that are critical for the formation of new electrostatic interactions.
Co-reporter:Rapole Srikanth, Jonathan Wilson, Colin S. Burns and Richard W. Vachet
Biochemistry 2008 Volume 47(Issue 35) pp:
Publication Date(Web):August 9, 2008
DOI:10.1021/bi800970m
While the Cu(II) binding sites of the prion protein have been well studied under Cu-saturation conditions, the identity of the residues involved in coordinating Cu(II) at low stoichiometries and the order in which the binding sites load with Cu(II) remain unresolved. In this study, we have used two mass spectrometry based methods to gather insight into Cu(II)-prion binding under different stoichiometric loadings of Cu(II). The first method uses metal-catalyzed oxidation reactions to site specifically modify the residues bound to Cu(II) in solution, and the second method determines Cu binding sites based on the protection of His from modification by diethyl pyrocarbonate when this residue binds Cu(II) in solution. For both methods, the residues that are labeled by these reactions can then be unambiguously identified using tandem mass spectrometry. Upon applying these two complementary methods to a construct of the prion protein that contains residues 23−28 and 57−98, several noteworthy observations are made. Coordination of Cu(II) by multiple His imidazoles is found at 1:1 and 1:2 PrP:Cu(II) ratios. Notably, there appear to be four to seven isomers of this multiple histidine coordination mode in the 1:1 complex. Furthermore, our data clearly show that His96 is the dominant Cu(II) binding ligand, as in every isomer His96 is bound to Cu(II). The individual octarepeat binding sites begin to fill at ratios of 1:3 PrP:Cu(II) with no clear preference for the order in which they load with Cu(II), although the His77 octarepeat appears to saturate last. The existence of several “degenerate” Cu binding modes at low PrP:Cu(II) ratios may allow it to more readily accept additional Cu(II) ions, thus allowing PrP to transition from a singly Cu(II) bound state to a multiply Cu(II) bound state as a function of cellular Cu(II) concentration.
Co-reporter:Juma D. Bridgewater, R. Srikanth, Jihyeon Lim, Richard W. Vachet
Journal of the American Society for Mass Spectrometry 2007 Volume 18(Issue 3) pp:553-562
Publication Date(Web):March 2007
DOI:10.1016/j.jasms.2006.11.001
Oxidative modifications to amino acid side chains can change the dissociation pathways of peptide ions, although these variations are most commonly observed when cysteine and methionine residues are oxidized. In this work we describe the very noticeable effect that oxidation of histidine residues can have on the dissociation patterns of peptide ions containing this residue. A common product ion spectral feature of doubly charged tryptic peptides is enhanced cleavage at the C-terminal side of histidine residues. This preferential cleavage arises as a result of the unique acid/base character of the imidazole side chain that initiates cleavage of a proximal peptide bond for ions in which the number of protons does not exceed the number of basic residues. We demonstrate here that this enhanced cleavage is eliminated when histidine is oxidized to 2-oxo-histidine because the proton affinity and nucleophilicity of the imidazole side chain are lowered. Furthermore, we find that oxidation of histidine to 2-oxo-histidine can cause the misassignment of oxidized residues when more than one oxidized isomer is simultaneously subjected to tandem mass spectrometry (MS/MS). These spectral misinterpretations can usually be avoided by using multiple stages of MS/MS (MSn) or by specially optimized liquid chromatographic separation conditions. When these approaches are not accessible or do not work, N-terminal derivatization with sulfobenzoic acid avoids the problem of mistakenly assigning oxidized residues.
Co-reporter:R. Srikanth;Jonathan Wilson
Journal of The American Society for Mass Spectrometry 2007 Volume 18( Issue 8) pp:1499-1506
Publication Date(Web):2007 August
DOI:10.1016/j.jasms.2007.05.011
Oxidative modifications to the side chains of sulfur-containing amino acids often limit the number of product ions formed during collision-induced dissociation (CID) and thus make it difficult to obtain sequence information for oxidized peptides. In this work, we demonstrate that electron-transfer dissociation (ETD) can be used to improve the sequence information obtained from peptides with oxidized cysteine and methionine residues. In contrast to CID, ETD is found to be much less sensitive to the side-chain chemistry, enabling extensive sequence information to be obtained in cases where CID fails to provide this information. These results indicate that ETD is a valuable technique for studying oxidatively modified peptides and proteins. In addition, we report a unique and very abundant product ion that is formed in the CID spectra of peptides having N-terminal cysteine sulfinic acid residues. The mechanism for this unique dissociation pathway involves a six-membered cyclic intermediate and leads to the facile loss of NH3 and SO2, which corresponds to a mass loss of 81 Da. While the facile nature of this dissociation pathway limits the sequence information present in CID spectra of peptides with N-terminal cysteine sulfinic acid residues, extensive sequence information for these peptides can be obtained with ETD.
Co-reporter:Juma D. Bridgewater, Richard W. Vachet
Analytical Biochemistry 2005 Volume 341(Issue 1) pp:122-130
Publication Date(Web):1 June 2005
DOI:10.1016/j.ab.2005.02.034
Further study has been made of metal-catalyzed oxidation (MCO) reactions and mass spectrometry as a method to determine the binding site of copper in metalloproteins. The role of ascorbate and a variety of oxidizing agents, including O2, H2O2, and S2O82−, have been investigated using Cu/Zn superoxide dismutase (SOD) as a model system. Ascorbate is found to play two competing roles in the MCO reactions. It reduces Cu(II), which initiates and maintains the generation of reactive oxygen species, and it scavenges radicals, which helps to localize oxidation products to amino acids near the metal center. An ascorbate concentration of 100 mM is found to be optimal with regard to localizing oxidation products to only the Cu-binding residues (His44, His46, His61, and His118) of Cu/Zn SOD. This concentration of ascorbate is very similar to the optimum concentration found in our previous studies of different Cu-binding proteins. Another notable result from this study is the observation that S2O82− is more effective as an oxidant than O2 or H2O2 in the MCO reactions. Because S2O82− is more stable in solution than H2O2, using it as an oxidizing agent results in much less nonspecific oxidation to the protein. The overall results of this study suggest that general MCO reaction conditions may exist for determining the metal-binding site of a wide range of Cu-binding proteins.
Co-reporter:Richard W Vachet, Myrasol B Callaway
Marine Chemistry 2003 Volume 82(1–2) pp:31-45
Publication Date(Web):June 2003
DOI:10.1016/S0304-4203(03)00047-1
The speciation of Cu(II) in marine waters is dominated by organic ligands, which have resisted detailed chemical characterization. In this work we have used immobilized-metal affinity chromatography (IMAC) to isolate Cu(II)-binding ligands from the Chesapeake Bay. We have then used high-performance size-exclusion chromatography (HPSEC) and mass spectrometry (MS) to gather information about the size distributions, molecular weights, and chemical functionality of the ligands isolated by IMAC. Results show that weaker-binding ligands have molecular weights that range from about 230 up to >20,000 Da. A portion of these weaker-binding ligands has molecular weight distributions that are consistent with humic substances. The molecular weight distribution of stronger-binding ligands is significantly more narrow with molecular weight values that are less than 1600 Da. Both HPSEC and MS show that the most abundant stronger-binding ligands have molecular weights around 270 Da. Furthermore, mass spectral analysis allows some empirical molecular formulas to be postulated for several ligands. These empirical formulas show that the ligands are abundant in nitrogen, oxygen, and sulfur functionality. In addition, consistency between MS data and data from HPSEC when peptides are used for the calibration combined with the low molecular weights and prevalence of nitrogen, oxygen, and sulfur functionality suggest that the stronger-binding ligands may have been produced directly by organisms in the water. It is not clear at this point, however, the degree to which the molecular information we have gathered represents the majority of the Cu(II)-binding ligands at our sampling site. Nonetheless, combining IMAC, HPSEC, and MS seems to be a promising approach for characterizing Cu(II)-binding ligands in natural waters.
Co-reporter:Marianny Y. Combariza, Richard W. Vachet
Journal of the American Society for Mass Spectrometry 2002 Volume 13(Issue 7) pp:813-825
Publication Date(Web):July 2002
DOI:10.1016/S1044-0305(02)00378-1
Using a modified quadrupole ion trap mass spectrometer, a series of metal complex ions have been reacted with acetonitrile in the gas phase. Careful control of the coordination number and the type of coordinating functionality in diethylenetriamine-substituted ligands enable the effects of the coordination sphere on metal complex reactivity to be examined. The association reaction kinetics of acetonitrile with these pentacoordinate complexes are followed in order to obtain information about the starting complexes and the reaction dynamics. The kinetics and thermodynamics of acetonitrile addition to the metal complex ions are strongly affected by the chemical environment around the metal center such that significant differences in reactivity are observed for Co(II) and Cu(II) complexes with various coordination spheres. When thiophene, furan, or benzene moieties are present in the coordination sphere of the complex, addition of two acetonitrile molecules is readily observed. In contrast, ligands with better σ donors react mainly to add one acetonitrile molecule. Among the ligands with good σ donors, a clear trend in reactivity is observed in which complexes with nitrogen-containing ligands are the least reactive, sulfur-containing complexes are more reactive, and oxygen-containing complexes are the most reactive. In general, equilibrium and reaction rate constants seem to be consistent with the hard and soft acid and base (HSAB) principle. Interestingly, the presence of certain groups (e.g., pyridine and imidazole) in the coordination sphere clearly can change the acid character of the metal as seen by their effect on the binding properties of other functional groups in the same ligand. Finally, we conclude that because complexes with different coordination spheres react to noticeably different extents, ion-molecule (I-M) reactions may be potentially useful for obtaining coordination structure information for transition metal complexes.
Co-reporter:R. Srikanth, Jonathan Wilson, Juma D. Bridgewater, Jason R. Numbers, Jihyeon Lim, Mark R. Olbris, Ali Kettani, Richard W. Vachet
Journal of the American Society for Mass Spectrometry (August 2007) Volume 18(Issue 8) pp:1499-1506
Publication Date(Web):1 August 2007
DOI:10.1016/j.jasms.2007.05.011
Oxidative modifications to the side chains of sulfur-containing amino acids often limit the number of product ions formed during collision-induced dissociation (CID) and thus make it difficult to obtain sequence information for oxidized peptides. In this work, we demonstrate that electron-transfer dissociation (ETD) can be used to improve the sequence information obtained from peptides with oxidized cysteine and methionine residues. In contrast to CID, ETD is found to be much less sensitive to the side-chain chemistry, enabling extensive sequence information to be obtained in cases where CID fails to provide this information. These results indicate that ETD is a valuable technique for studying oxidatively modified peptides and proteins. In addition, we report a unique and very abundant product ion that is formed in the CID spectra of peptides having N-terminal cysteine sulfinic acid residues. The mechanism for this unique dissociation pathway involves a six-membered cyclic intermediate and leads to the facile loss of NH3 and SO2, which corresponds to a mass loss of 81 Da. While the facile nature of this dissociation pathway limits the sequence information present in CID spectra of peptides with N-terminal cysteine sulfinic acid residues, extensive sequence information for these peptides can be obtained with ETD.
.jpg)

![BENZENE, 1-[(10-BROMODECYL)OXY]-4-METHYL-](http://img.cochemist.com/ccimg/90600/90504-88-4.png)
![BENZENE, 1-[(10-BROMODECYL)OXY]-4-METHYL-](http://img.cochemist.com/ccimg/90600/90504-88-4_b.png)
![Benzene, 1-[(10-bromodecyl)oxy]-4-nitro-](http://img.cochemist.com/ccimg/87500/87477-66-5.png)
![Benzene, 1-[(10-bromodecyl)oxy]-4-nitro-](http://img.cochemist.com/ccimg/87500/87477-66-5_b.png)


![Benzene,[(10-bromodecyl)oxy]-](http://img.cochemist.com/ccimg/2100/2033-87-6.png)
![Benzene,[(10-bromodecyl)oxy]-](http://img.cochemist.com/ccimg/2100/2033-87-6_b.png)







































































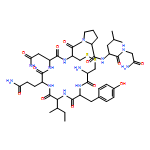
![2-[[1-[2-[[2-[(2-amino-3-phenylpropanoyl)amino]-3-(4-hydroxyphenyl)propanoyl]amino]acetyl]pyrrolidine-2-carbonyl]amino]-3-methylbutanoic Acid](http://img.cochemist.com/ccimg/85700/85679-70-5.png)
![2-[[1-[2-[[2-[(2-amino-3-phenylpropanoyl)amino]-3-(4-hydroxyphenyl)propanoyl]amino]acetyl]pyrrolidine-2-carbonyl]amino]-3-methylbutanoic Acid](http://img.cochemist.com/ccimg/85700/85679-70-5_b.png)

![Phosphonothioic acid,P-methyl-, S-[2-[bis(1-methylethyl)amino]ethyl] O-ethyl ester](http://img.cochemist.com/ccimg/50800/50782-69-9.png)
![Phosphonothioic acid,P-methyl-, S-[2-[bis(1-methylethyl)amino]ethyl] O-ethyl ester](http://img.cochemist.com/ccimg/50800/50782-69-9_b.png)





































![2-[(2E)-2-(2-CHLOROBENZYLIDENE)HYDRAZINO]-5-NITROPYRIDINE](http://img.cochemist.com/ccimg/100/77-81-6.png)
![2-[(2E)-2-(2-CHLOROBENZYLIDENE)HYDRAZINO]-5-NITROPYRIDINE](http://img.cochemist.com/ccimg/100/77-81-6_b.png)