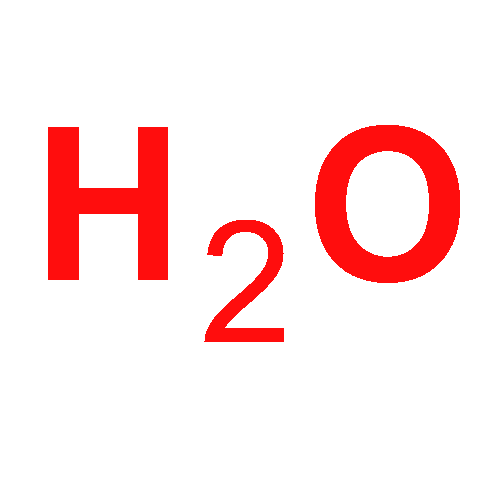
Co-reporter:Bess Vlaisavljevich and Toru Shiozaki
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 8) pp:3781-3787
Publication Date(Web):July 7, 2016
DOI:10.1021/acs.jctc.6b00572
We report the development of the theory and computer program for analytical nuclear energy gradients for (extended) multistate complete active space perturbation theory (CASPT2) with full internal contraction. The vertical shifts are also considered in this work. This is an extension of the fully internally contracted CASPT2 nuclear gradient program recently developed for a state-specific variant by us [MacLeod and Shiozaki, J. Chem. Phys. 2015, 142, 051103]; in this extension, the so-called λ equation is solved to account for the variation of the multistate CASPT2 energies with respect to the change in the amplitudes obtained in the preceding state-specific CASPT2 calculations, and the Z vector equations are modified accordingly. The program is parallelized using the MPI3 remote memory access protocol that allows us to perform efficient one-sided communication. The optimized geometries of the ground and excited states of a copper corrole and benzophenone are presented as numerical examples. The code is publicly available under the GNU General Public License.
Co-reporter:Toru Shiozaki and Takeshi Yanai
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 9) pp:4347-4351
Publication Date(Web):August 1, 2016
DOI:10.1021/acs.jctc.6b00646
We present an accurate method for calculating hyperfine coupling constants (HFCCs) based on the complete active space second-order perturbation theory (CASPT2) with full internal contraction. The HFCCs are computed as a first-order property using the relaxed CASPT2 spin-density matrix that takes into account orbital and configurational relaxation due to dynamical electron correlation. The first-order unrelaxed spin-density matrix is calculated from one- and two-body spin-free counterparts that are readily available in the CASPT2 nuclear gradient program [M. K. MacLeod and T. Shiozaki, J. Chem. Phys. 142, 051103 (2015)], whereas the second-order part is computed directly using the newly extended automatic code generator. The relaxation contribution is then calculated from the so-called Z-vectors that are available in the CASPT2 nuclear gradient program. Numerical results are presented for the CN and AlO radicals, for which the CASPT2 values are comparable (or, even superior in some cases) to the ones computed by the coupled-cluster and density matrix renormalization group methods. The HFCCs for the hexaaqua complexes with VII, CrIII, and MnII are also presented to demonstrate the accuracy and efficiency of our code.
Co-reporter:Inkoo Kim, Shane M. Parker, and Toru Shiozaki
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 8) pp:3636-3642
Publication Date(Web):July 16, 2015
DOI:10.1021/acs.jctc.5b00429
We report the derivation and implementation of orbital optimization algorithms for the active space decomposition (ASD) model, which are extensions of complete active space self-consistent field (CASSCF) and its occupation-restricted variants in the conventional multiconfiguration electronic-structure theory. Orbital rotations between active subspaces are included in the optimization, which allows us to unambiguously partition the active space into subspaces, enabling application of ASD to electron and exciton dynamics in covalently linked chromophores. One- and two-particle reduced density matrices, which are required for evaluation of orbital gradient and approximate Hessian elements, are computed from the intermediate tensors in the ASD energy evaluation. Numerical results on 4-(2-naphthylmethyl)-benzaldehyde and [36]cyclophane and model Hamiltonian analyses of triplet energy transfer processes in the Closs systems are presented. Furthermore, model Hamiltonians for hole and electron transfer processes in anti-[2.2](1,4)pentacenophane are studied using an occupation-restricted variant.
Co-reporter:Toru Shiozaki and Wataru Mizukami
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 10) pp:4733-4739
Publication Date(Web):September 14, 2015
DOI:10.1021/acs.jctc.5b00754
We report internally contracted relativistic multireference configuration interaction (ic-MRCI), complete active space second-order perturbation (CASPT2), and strongly contracted n-electron valence state perturbation theory (NEVPT2) on the basis of the four-component Dirac Hamiltonian, enabling accurate simulations of relativistic, quasi-degenerate electronic structure of molecules containing transition-metal and heavy elements. Our derivation and implementation of ic-MRCI and CASPT2 are based on an automatic code generator that translates second-quantized ansätze to tensor-based equations, and to efficient computer code. NEVPT2 is derived and implemented manually. The rovibrational transition energies and absorption spectra of HI and TlH are presented to demonstrate the accuracy of these methods.
Co-reporter:Ryan D. Reynolds and Toru Shiozaki
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 22) pp:14280-14283
Publication Date(Web):07 Oct 2014
DOI:10.1039/C4CP04027A
We present a gauge-invariant implementation of the four-component Dirac–Hartree–Fock method for simulating the electronic structure of heavy element complexes in magnetic fields. The additional cost associated with the magnetic field is shown to be only 10–13% of that at zero field. The Dirac–Hartree–Fock wave function is constructed from gauge-including atomic orbitals. The so-called restricted magnetic balance is used to generate 2-spinor basis functions for the small component. The molecular integrals for the Coulomb and Gaunt interactions are computed using density fitting. Our efficient, parallel implementation allows for simulating the electronic structure of molecules containing more than 100 atoms with a few heavy elements under magnetic fields.
Co-reporter:Shane M. Parker and Toru Shiozaki
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 9) pp:3738-3744
Publication Date(Web):July 15, 2014
DOI:10.1021/ct5004753
We present ab initio theory and efficient algorithms for computing model Hamiltonians of excited-state dynamics in the quasi-diabatic representation. The method is based on a recently developed multiconfiguration electronic structure method, called the active space decomposition method (ASD), in which quasi-diabatic basis states are constructed from physical fragment states. An efficient tree-based algorithm is presented for computing and reusing intermediate tensors appearing in the ASD model. Parallel scalability and wall times are reported to attest the efficiency of our program. Applications to electron, hole, and triplet energy transfers in molecular dimers are presented, demonstrating its versatility.
Co-reporter:Shane M. Parker ; Tamar Seideman ; Mark A. Ratner
The Journal of Physical Chemistry C 2014 Volume 118(Issue 24) pp:12700-12705
Publication Date(Web):May 28, 2014
DOI:10.1021/jp505082a
We present an approach to accurately construct the few-state model Hamiltonians for singlet fission processes on the basis of an ab initio electronic structure method tailored to dimer wave functions, called an active space decomposition strategy. In this method, the electronic structure of molecular dimers is expressed in terms of a linear combination of products of monomer states. We apply this method to tetracene and pentacene, using monomer wave functions computed by the restricted active space (RAS) method. Near-exact wave functions are computed for π-electrons of dimers that contain up to 7 × 1012 electronic configurations. Our product ansatz preserves the diabatic picture of the minimal dimer model, allowing us to accurately identify model Hamiltonians. The wave functions obtained from the model Hamiltonians account for more than 99% of the total wave functions. The resulting model Hamiltonians are shown to be converged with respect to all the parameters in the model, and corroborate previously reported coupling strengths.
Co-reporter:Toru Shiozaki
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 10) pp:4300-4303
Publication Date(Web):September 11, 2013
DOI:10.1021/ct400719d
An efficient algorithm is presented for evaluating the analytical nuclear gradients of density-fitted four-component relativistic Dirac–Fock theory as an initial step toward realizing large-scale geometry optimization of heavy-element complexes. Our algorithm employs kinetically balanced 2-spinor basis functions for the small components. The computational cost of nuclear gradient evaluation is found to be smaller than that of a Dirac–Fock self-consistent iteration. Timing data are presented for Ir(ppy)3 (61 atoms) using a double-ζ basis set.
Co-reporter:Toru Shiozaki, Clemens Woywod and Hans-Joachim Werner
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 1) pp:262-269
Publication Date(Web):25 Oct 2012
DOI:10.1039/C2CP43381H
We demonstrate that the recently developed extended multi-state complete active space second-order perturbation theory (XMS-CASPT2) [Shiozaki et al., J. Chem. Phys., 2011, 135, 081106] provides qualitatively correct potential energy surfaces for low-lying excited singlet states of pyrazine, while the potential energy surfaces of the standard MS-CASPT2 methods are ill-behaved near the crossing point of two reference potential energy surfaces. The XMS-CASPT2 method is based on the extended multi-configuration quasi-degenerate perturbation theory proposed earlier by Granovsky [J. Chem. Phys., 2011, 134, 214113]. We show that the conical intersection at the XMS-CASPT2 level can be described without artifacts if the entire method is invariant with respect to any unitary rotations of the reference functions. The photoabsorption spectra of the 11B3u and 11B2u states of pyrazine are simulated, based on a vibronic-coupling model Hamiltonian. The XMS-CASPT2 spectrum of the 11B3u band is found to be comparable to the one computed by a more expensive multireference configuration interaction (MRCI) method, while the XMS-CASPT2 simulation of the 11B2u band is slightly inferior to the MRCI one.
Co-reporter:Toru Shiozaki, Clemens Woywod and Hans-Joachim Werner
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 1) pp:NaN269-269
Publication Date(Web):2012/10/25
DOI:10.1039/C2CP43381H
We demonstrate that the recently developed extended multi-state complete active space second-order perturbation theory (XMS-CASPT2) [Shiozaki et al., J. Chem. Phys., 2011, 135, 081106] provides qualitatively correct potential energy surfaces for low-lying excited singlet states of pyrazine, while the potential energy surfaces of the standard MS-CASPT2 methods are ill-behaved near the crossing point of two reference potential energy surfaces. The XMS-CASPT2 method is based on the extended multi-configuration quasi-degenerate perturbation theory proposed earlier by Granovsky [J. Chem. Phys., 2011, 134, 214113]. We show that the conical intersection at the XMS-CASPT2 level can be described without artifacts if the entire method is invariant with respect to any unitary rotations of the reference functions. The photoabsorption spectra of the 11B3u and 11B2u states of pyrazine are simulated, based on a vibronic-coupling model Hamiltonian. The XMS-CASPT2 spectrum of the 11B3u band is found to be comparable to the one computed by a more expensive multireference configuration interaction (MRCI) method, while the XMS-CASPT2 simulation of the 11B2u band is slightly inferior to the MRCI one.
Co-reporter:Ryan D. Reynolds and Toru Shiozaki
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 22) pp:NaN14283-14283
Publication Date(Web):2014/10/07
DOI:10.1039/C4CP04027A
We present a gauge-invariant implementation of the four-component Dirac–Hartree–Fock method for simulating the electronic structure of heavy element complexes in magnetic fields. The additional cost associated with the magnetic field is shown to be only 10–13% of that at zero field. The Dirac–Hartree–Fock wave function is constructed from gauge-including atomic orbitals. The so-called restricted magnetic balance is used to generate 2-spinor basis functions for the small component. The molecular integrals for the Coulomb and Gaunt interactions are computed using density fitting. Our efficient, parallel implementation allows for simulating the electronic structure of molecules containing more than 100 atoms with a few heavy elements under magnetic fields.
.jpg)