Co-reporter:Yong WANG, Li WU, Jin-Ling XU, Shui-Ming LI, ... Liang JIANG
Chinese Journal of Analytical Chemistry 2017 Volume 45, Issue 10(Volume 45, Issue 10) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/S1872-2040(17)61044-2
AbstractAs an extension of proteomics, peptidomics has been widely used in medical and biological researches. However, the effect of reproducibility of identification method on peptidome is not clears. In this work, urine sample of a healthy people was analyzed for seven times in parallel by nano-liquid chromatography-high resolution tandem mass spectrometry (Nano-LC-MS/MS). To illustrate the variability and stability among these experiments, the number of spectra, the utilization of total spectra, the number of identified peptides, the number of proteins, and the ionic strength and retention time of peptides have been counted and compared. The average number of peptides was 208 and the standard deviation was 38.7. After all of data were combined, 426 peptides belonging to 114 proteins were obtained, while only 109 peptides coming from 35 proteins were identified in each experiment, indicating that there was both an randomness and a relative stability for LC-MS analysis. Increasing the number of parallel experiments would expand the data set of polypeptides, but the rate of increase would decrease after 3 or more measurements. In comparison with peptides, the results of peptidomics were more stable at protein level, indicating that proteins were more robust peptidomics biomarker than peptides.The urine sample of a healthy people was analyzed for 7 times in parallel by using nano-liquid chromatography-high-resolution tandem mass spectrometry to explore the reproducibility of peptidomics technique. The data set of peptidomics was expanded with the increase of the number of parallel experiments, but 109 peptides coming from 35 high abundant proteins were identified in each experiment. These results showed that proteins were more robustly peptidomics biomarker than the peptides.Download high-res image (92KB)Download full-size image
Co-reporter:Hao Wang;Xiaoyu Hong;Shuiming Li
Journal of Molecular Neuroscience 2017 Volume 63( Issue 2) pp:243-253
Publication Date(Web):21 September 2017
DOI:10.1007/s12031-017-0975-0
Protein synthesis has been reported to be impaired in early-stage Alzheimer’s disease (AD). Previously, we found that oxygen supplementation improved cognitive function and reduced mitochondrial damage in AD model mice. In the present study, we examined the effects of supplemental oxygen treatment on protein synthesis and oxidative damage. The synthesis of numerous proteins involved in mRNA splicing, transcription regulation, and translation was found to be significantly upregulated in cortex tissues of AD model mice given a supplemental oxygen treatment (OT group), relative to those of non-treated control AD model mice (Ctrl group), suggesting that impairment in protein synthesis may be alleviated by increased oxygen inhalation. Methionine oxidation and oxidation levels in general were similar between the OT and Ctrl groups, indicating that the oxygen supplementation treatment did not cause increases in peptide oxidation levels. On the contrary, the OT group exhibited upregulation of several proteins associated with antioxidant defense. These results support further exploration into the development of supplementary oxygen treatment as a potential therapy for AD.
Co-reporter:Xiao-Yu HONG, Shui-Ming LI, Yong WANG
Chinese Journal of Analytical Chemistry 2016 Volume 44(Issue 5) pp:716-722
Publication Date(Web):May 2016
DOI:10.1016/S1872-2040(16)60931-3
Proteomics is an emerging field branching from proteomics that targets endogenous peptides of a whole organism or a subsystem. Many peptides in body fluids including urine are biomarkers with higher clinical sensitivity and specificity. In this study, we separated and enriched these peptides in human urine by LaPO4-graphene oxide composite nanomaterial, then identified and analyzed them by nano liquid chromatography-high resolution tandem mass spectrometry. In single urine sample, we identified 790 peptides which belonged to 123 proteins and some post-translation modifications including oxidation, phosphorylation, deamidated etc. There existed a series of peptide ladders in urine peptidomic. Finally, the string analysis at protein level revealed strong interaction among these proteins to which the identified peptides belong. This method can provide support for finding the biomarkers of disease in urine.Here, we separated and enriched the low abundance peptides in human urine by LaGM composites, then identified and analyzed by nanoLC-MS/MS. We identified 790 peptides arised from 123 proteins and PTMs. Finally, the analysis at protein level revealed strong interaction among these proteins. This method can provide support for urine peptidomics analysis.
Co-reporter:Yong WANG, Shui-Ming LI, Man-Wen HE
Chinese Journal of Analytical Chemistry 2014 Volume 42(Issue 7) pp:1010-1016
Publication Date(Web):July 2014
DOI:10.1016/S1872-2040(14)60752-0
One of significant characteristics of matrix-assisted laser desorption/ionization tandem time-of-light mass spectrometry (MALDI-TOF/TOF-MS) high-energy collision induced dissociation (CID) is to produce abundant immonium (IM) ions that can offer a wealth of information for peptide composition. However, MALDI-TOF/TOF is generally used for routine protein identification based on database search or de novo sequencing combined with chemical derivation. Consequently, the characteristics of IM ions may not be fully explored and utilized. Here, a total of 239 MS/MS spectra were used to explore the fragmentation features of IM ions with MALDI TOF/TOF spectrometry and their application for peptide identification. IM ion signals for 14 kinds of amino acids including His with a positive rate of more than 50% were observed. The results indicated that the chemical nature of the amino acids and position effect were two main factors that affect the intensity of fragment ions. In addition, false positive IM ions were mainly derived from Arg, Lys, Leu and Ile residues or mixture peptides. Besides the compositional information, partial sequence information was also obtained by comparison of the relative intensity of IM ions. These findings are helpful when performing manual interpretations and can help improving current peptide search algorithms.Besides a nearly complete series of b- and y- ion, another advantage of high energy CID performed on TOF/TOF is that the method can produce immonium ions, which can be used to distinguish isobaric peptides such as DGEAAENTDAQK and DSVAAENTDAQK. A strong Val IM ion was observed at m/z 72, which indicated that there are more than one Valine residues in sequence or it occupies a favorable position effect.

![N-[3,5-Bis(trifluoromethyl)phenyl]-N′-[(8a,9S)-10,11-dihydro-6′-methoxy-9-cinchonanyl]thiourea](/data/chemimg/1588800/852913-19-0.png)
![N-[3,5-Bis(trifluoromethyl)phenyl]-N′-[(8a,9S)-10,11-dihydro-6′-methoxy-9-cinchonanyl]thiourea](/data/chemimg/1588800/852913-19-0_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)


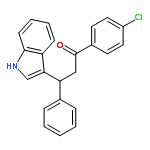
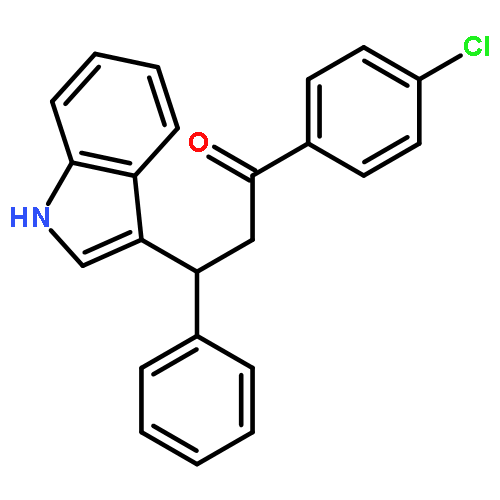

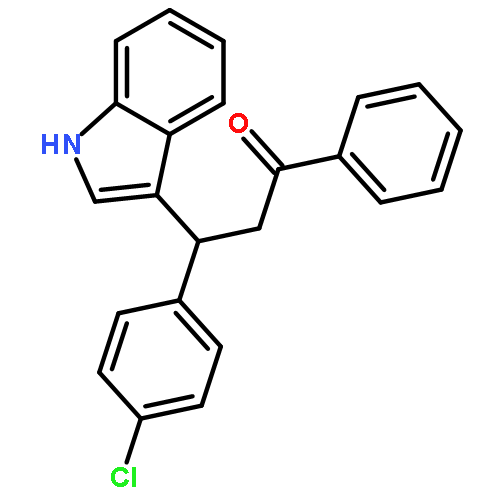

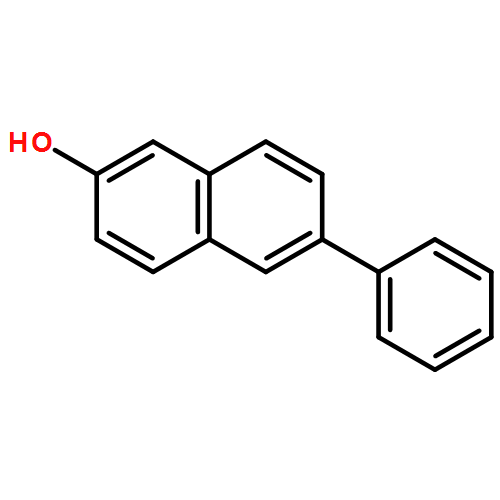
















![Benzene, [(butylthio)ethynyl]-](http://img.cochemist.com/ccimg/127100/127060-60-0.png)
![Benzene, [(butylthio)ethynyl]-](http://img.cochemist.com/ccimg/127100/127060-60-0_b.png)
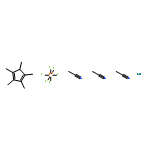
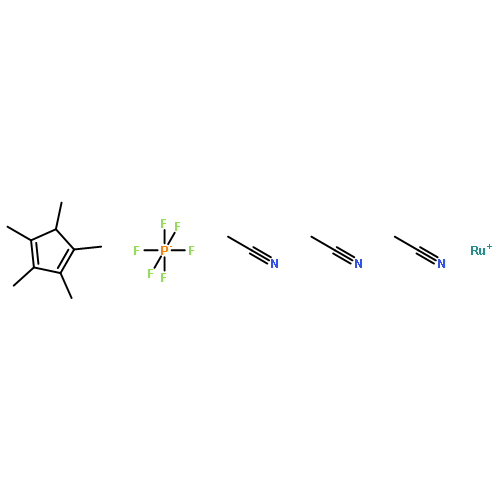
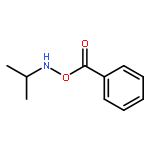
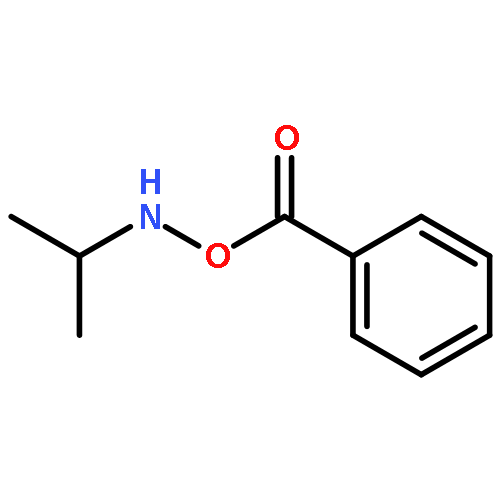


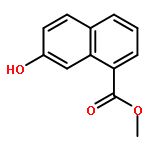
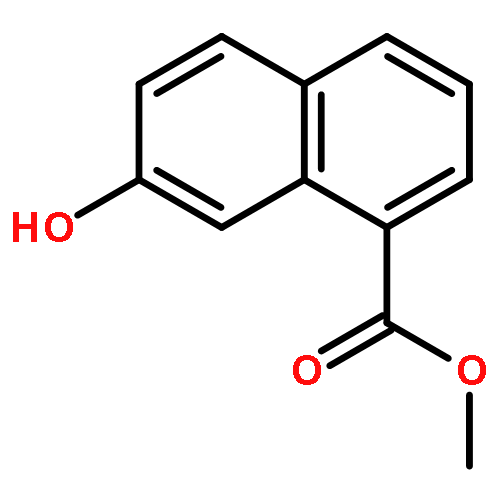


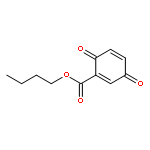
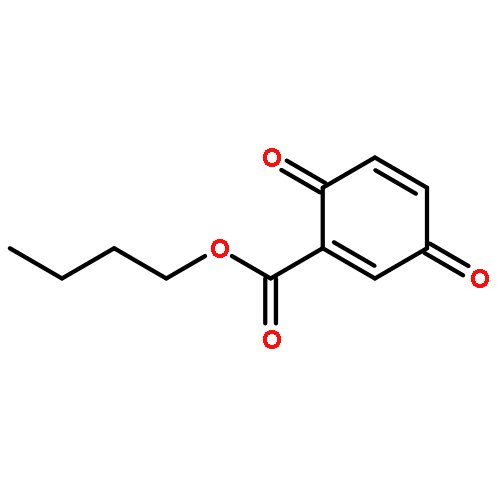























![SODIUM {[2-METHOXY-5-({[(E)-2-(2,4,6-TRIMETHOXYPHENYL)VINYL]SULFO<WBR />NYL}METHYL)PHENYL]AMINO}ACETATE](http://img.cochemist.com/ccimg/4000/3943-91-7.png)
![SODIUM {[2-METHOXY-5-({[(E)-2-(2,4,6-TRIMETHOXYPHENYL)VINYL]SULFO<WBR />NYL}METHYL)PHENYL]AMINO}ACETATE](http://img.cochemist.com/ccimg/4000/3943-91-7_b.png)