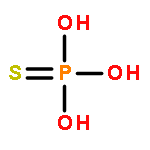
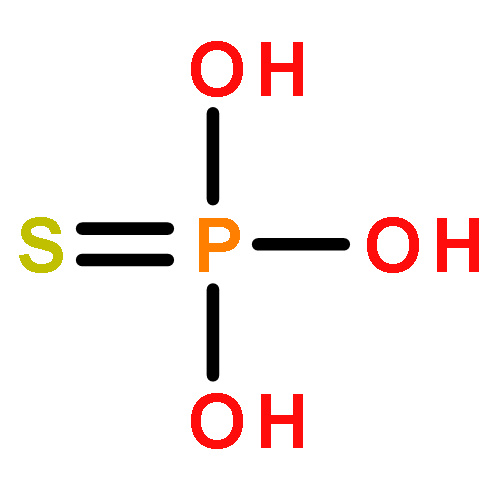
Co-reporter:Zhongli Wang, Wenjuan Xie, Mingyan Zhu, Huchen Zhou
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 17(Issue 17) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.bmcl.2017.07.059
The dynamic modification of proteins with ubiquitin plays crucial roles in major celluar functions, and is associated with a number of pathological conditions. Ubiquitin-specific proteases (USPs) cleave ubiquitin from substrate proteins, and rescue them from proteasomal degradation. Among them, USP2 is overexpressed and plays important roles in various cancers including prostate cancer. Thus, it represents an attractive target for drug discovery. In order to develop potent and selective USP2 inhibitors, a highly reliable assay is needed for in-depth structure-activity relationship study. We report the cloning, expression, and purification of USP2 and UBA52, and the development of a highly reliable assay based on readily available SDS-PAGE-Coomassie systeme using UBA52 as the substrate protein. A number of effective USP2 inhibitors were also identified using this assay.Download high-res image (100KB)Download full-size image
Co-reporter:Yaxue Zhao, Zhongli Wang, Jianchen Zhang, Huchen Zhou
European Journal of Medicinal Chemistry 2016 Volume 122() pp:178-184
Publication Date(Web):21 October 2016
DOI:10.1016/j.ejmech.2016.06.018
•Eleven new scaffolds as SENP1 inhibitors discovered through in silico screening.•New SENP1 inhibitors synthesized and a preliminary SAR discussed.•Compound 13m showed an IC50 as low as 3.5 μM.The small ubiquitin-related modifier (SUMO)-specific proteases (SENPs) catalyze the deconjugation of SUMO from their substrate proteins. SENP1 which is the most studied isoform is closely related to many cancers such as prostate cancer and colon cancer, thus representing a potential therapeutic target for cancer treatment. In the present study, we identified eleven SENP1 inhibitors representing a variety of scaffolds through in silico screening. Based on these scaffolds, a series of new compounds were designed and synthesized in order to improve their SENP1 inhibitory potency. As a result, compounds with IC50 as low as 3.5 μM (compound 13m) were obtained and a preliminary structure-activity relationship was discussed.
Co-reporter:Wenjuan Xie, Zhongli Wang, Jianchen Zhang, Lie Wang, Yaxue Zhao, Huchen Zhou
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 9) pp:2124-2128
Publication Date(Web):1 May 2016
DOI:10.1016/j.bmcl.2016.03.080
SUMOylation, as a post-translational modification of proteins, plays essential regulatory roles in a variety of pathological conditions. In the dynamic process of SUMOylation and deSUMOylation, SENPs (SUMO-specific proteases), in charge of deconjugation of SUMO (small ubiquitin-related modifier) from substrate proteins, have recently been found to be potential therapeutic targets for cancer treatment. A reliable and practical assay is much needed to accelerate the discovery of SENPs inhibitors. We established a quantitative assay based on readily available SDS–PAGE-Coomassie system using RanGAP-SUMO as the substrate, thus avoiding the use of expensive fluorescent dyes or the error-prone fluorescent reporter. Its reproducibility and reliability were also evaluated in this report.
Co-reporter:Puhua Wu, Jiong Zhang, Qingqing Meng, Bakela Nare, Robert T. Jacobs, Huchen Zhou
European Journal of Medicinal Chemistry 2014 Volume 81() pp:59-75
Publication Date(Web):23 June 2014
DOI:10.1016/j.ejmech.2014.04.079
•A new class of benzoxaboroles, pyrrolobenzoxaboroles, were synthesized as antitrypanosomal agents.•The 3′-acylpyrrolyl derivatives gave the most favorable activity with IC50 values as low as 0.03 μg/mL.•Three of the lead compounds were demonstrated to cure the parasitic infection in an acute infection murine model.Human African trypanosomiasis is a fatal parasitic infection caused by the protozoan Trypanosoma brucei. The development of novel antitrypanosomal agents is urgently needed. Here we report the synthesis and structure–activity relationship of a new class of benzoxaboroles as antitrypanosomal agents. These compounds showed antiparasitic IC50 values ranging from 4.02 to 0.03 μg/mL and satisfactory cytotoxicity profile. Three of the lead compounds were demonstrated to cure the parasitic infection in a murine acute infection model. The structure–activity relationship of the pyrrolobenzoxaboroles are also discussed.A new class of benzoxaboroles were synthesized as antitrypanosomal agents and showed IC50 as low as 0.03 μg/mL. Three of the lead compounds eliminated parasitic infection in a murine model.
Co-reporter:Fenglong Zhang, Jin Du, Qing Wang, Qinghua Hu, Jiong Zhang, Dazhong Ding, Yaxue Zhao, Fei Yang, Enduo Wang and Huchen Zhou
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 32) pp:5310-5324
Publication Date(Web):19 Jun 2013
DOI:10.1039/C3OB40236C
Human African trypanosomiasis (HAT) is one of the most neglected diseases in the tropic regions, which is fatal if not treated in time. There is an urgent need for new therapeutics, especially those in new chemical classes. Leucyl-tRNA synthetase (LeuRS) has been paid much attention as a recently clinically validated antimicrobial target. Our group has previously reported T. brucei LeuRS (TbLeuRS) inhibitors, including benzoxaboroles targeting the editing site and pyrrolinones targeting the synthetic site. Here we report the discovery of N-(4-sulfamoylphenyl)thioureas as a new class of TbLeuRS inhibitors. The R1 and R2 groups, reminiscent of the leucyl and adenyl regions of aa-AMP and aa-AMS, were optimized to result in a significant 13-fold increase of inhibitory activity (compound 19, IC50 = 13.7 μM). Aided by ligand–protein docking, the 1,3-substitution at the central phenyl ring was predicted and proved to give significantly improved activity (59, IC50 = 1.1 μM). This work provided a new scaffold for the exploration of novel inhibitors against TbLeuRS, which may become potential therapeutics for the treatment of HAT.
Co-reporter:Yaxue Zhao, Feng Zhou, Huchen Zhou and Haibin Su
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 5) pp:1690-1698
Publication Date(Web):28 Nov 2012
DOI:10.1039/C2CP42830J
The bonding characteristics in cysteine–gold cluster complexes represented by thiolate (Aun·CysS (n = 1, 3, 5, 7)) and thiol (Aun·CysSH (n = 2, 4, 6, 8)) is investigated by density functional theory with 6-31G(d,p) and Lanl2DZ hybrid basis sets. The complexes exhibit very different bonding characteristic between these two forms. In the Aun·CysS complexes, the charge transfers from gold clusters to sulfur atoms. The number of S–Au bonds in the Aun·CysS complexes evolves from one to two when n is greater than three. For n equals three, i.e. Au3·CysS, its ground state only has one S–Au bond. While the only S–Au bond in Au1·CysS is mainly covalent, the nature of the S–Au bond in other thiolates is featured with the combination of covalent and donor–acceptor interactions. In particular, one stable isomer of Au3·CysS with two S–Au bonds, which is 2 kcal mol−1 higher in energy than the corresponding ground state, consists of one covalent and one donor–acceptor S–Au bond explicitly. Moreover, the localized three center two electron bonds are formed within the Au clusters, which facilitates the formation of the two S–Au bonds in Au5·CysS and Au7·CysS complexes. In the Aun·CysSH complexes, the donor–acceptor interaction prevails in the Au–SH bond by transferring lone pair electrons from the sulfur atom to the adjacent gold atom. Interestingly, the orbital with much more 6s-component in Au4·CysSH enhances the donor–acceptor bonding character, thus yields the strongest bonding among all the Aun·CysSH complexes studied in this paper. In general, the bonding strength between gold clusters and cysteine is positively correlated with the S–Au overlap-weighted bond order, but negatively correlated with the S–Au bond length. Lastly, the covalent and donor–acceptor S–Au bond strength is computed to be 48 and 18 kcal mol−1, respectively.
Co-reporter:Jiong Zhang;MingYan Zhu;YiNan Lin
Science China Chemistry 2013 Volume 56( Issue 10) pp:1372-1381
Publication Date(Web):2013 October
DOI:10.1007/s11426-013-4981-y
Benzoxaborole, as a versatile scaffold, plays important roles in organic synthesis, molecular recognition and supramolecular chemistry. It is also a privileged structure in medicinal chemistry due to its desirable physicochemical and drug-like properties. Recently, benzoxaboroles were widely applied as antifungal, antibacterial, antiviral, anti-parasite, and anti-inflammatory agents. This review covers the properties, synthetic methods and applications of benzoxaboroles in medicinal chemistry.
Co-reporter:Amy Friesland;Yaxue Zhao;Qun Lu;Lie Wang;Yan-Hua Chen
PNAS 2013 Volume 110 (Issue 4 ) pp:1261-1266
Publication Date(Web):2013-01-22
DOI:10.1073/pnas.1116051110
Signaling through the Rho family of small GTPases has been intensely investigated for its crucial roles in a wide variety
of human diseases. Although RhoA and Rac1 signaling pathways are frequently exploited with the aid of effective small molecule
modulators, studies of the Cdc42 subclass have lagged because of a lack of such means. We have applied high-throughput in silico screening and identified compounds that are able to fit into the surface groove of Cdc42, which is critical for guanine nucleotide
exchange factor binding. Based on the interaction between Cdc42 and intersectin (ITSN), a specific Cdc42 guanine nucleotide
exchange factor, we discovered compounds that rendered ITSN-like interactions in the binding pocket. By using in vitro binding
and imaging as well as biochemical and cell-based assays, we demonstrated that ZCL278 has emerged as a selective Cdc42 small
molecule modulator that directly binds to Cdc42 and inhibits its functions. In Swiss 3T3 fibroblast cultures, ZCL278 abolished
microspike formation and disrupted GM130-docked Golgi structures, two of the most prominent Cdc42-mediated subcellular events.
ZCL278 reduces the perinuclear accumulation of active Cdc42 in contrast to NSC23766, a selective Rac inhibitor. ZCL278 suppresses
Cdc42-mediated neuronal branching and growth cone dynamics as well as actin-based motility and migration in a metastatic prostate
cancer cell line (i.e., PC-3) without disrupting cell viability. Thus, ZCL278 is a small molecule that specifically targets
Cdc42–ITSN interaction and inhibits Cdc42-mediated cellular processes, thus providing a powerful tool for research of Cdc42
subclass of Rho GTPases in human pathogenesis, such as those of cancer and neurological disorders.
Co-reporter:Zhitao Qiao ; Qi Wang ; Fenglong Zhang ; Zhongli Wang ; Tana Bowling ; Bakela Nare ; Robert T. Jacobs ; Jiong Zhang ; Dazhong Ding ; Yangang Liu
Journal of Medicinal Chemistry 2012 Volume 55(Issue 7) pp:3553-3557
Publication Date(Web):February 23, 2012
DOI:10.1021/jm2012408
We report the novel chalcone–benzoxaborole hybrids and their structure–activity relationship againstTrypanosoma brucei parasites. The 4-NH2 derivative 29 and 3-OMe derivative 43 were found to have excellent potency. The synergistic 4-NH2-3-OMe compound 49 showed an IC50 of 0.010 μg/mL and resulted in 100% survival and zero parasitemia in a murine infection model, which represents one of the most potent compounds discovered to date from the benzoxaborole class that inhibit T. brucei growth.
Co-reporter:Yaxue Zhao, Qing Wang, Qingqing Meng, Dazhong Ding, Huaiyu Yang, Guangwei Gao, Dawei Li, Weiliang Zhu, Huchen Zhou
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 3) pp:1240-1250
Publication Date(Web):1 February 2012
DOI:10.1016/j.bmc.2011.12.035
Human African trypanosomiasis (HAT), caused by the protozoan parasite Trypanosoma brucei, is a neglected fatal disease. Leucyl-tRNA synthetase (LeuRS), which has been successfully applied in the development of antifungal agent, represents a potential antiprotozoal drug target. In this study, a 3D model of T. brucei LeuRS (TbLeuRS) synthetic active site was constructed and subjected to virtual screening using a combination of pharmacophore- and docking-based methods. A new 2-pyrrolinone scaffold was discovered and the structure–activity relationship (SAR) studies aided by the docking model and organic synthesis were carried out. Compounds with various substituents on R1, R2 and R3 were synthesized and their SAR was discussed.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Dazhong Ding ; Qingqing Meng ; Guangwei Gao ; Yaxue Zhao ; Qing Wang ; Bakela Nare ; Robert Jacobs ; Fernando Rock ; Michael R. K. Alley ; Jacob J. Plattner ; Guoqiang Chen ; Dawei Li
Journal of Medicinal Chemistry 2011 Volume 54(Issue 5) pp:1276-1287
Publication Date(Web):February 15, 2011
DOI:10.1021/jm101225g
African trypanosomiasis, caused by the proto zoal pathogen Trypanosoma brucei (T. brucei), is one of the most neglected tropical diseases that are in great need of new drugs. We report the design and synthesis of T. brucei leucyl-tRNA synthetase (TbLeuRS) inhibitors and their structure−activity relationship. Benzoxaborole was used as the core structure and C(6) was modified to achieve improved affinity based on docking results that showed further binding space at this position. Indeed, compounds with C(7) substitutions showed diminished activity due to clash with the eukaryote specific I4ae helix while substitutions at C(6) gave enhanced affinity. TbLeuRS inhibitors with IC50 as low as 1.6 μM were discovered, and the structure−activity relationship was discussed. The most potent enzyme inhibitors also showed excellent T. brucei parasite growth inhibition activity. This is the first time that TbLeuRS inhibitors are reported, and this study suggests that leucyl-tRNA synthetase (LeuRS) could be a potential target for antiparasitic drug development.
Co-reporter:Mingxuan Wu, Qingqing Meng, Min Ge, Linquan Bai, Huchen Zhou
Tetrahedron Letters 2011 Volume 52(Issue 44) pp:5799-5801
Publication Date(Web):2 November 2011
DOI:10.1016/j.tetlet.2011.08.132
The synthesis and characterization of highly challenging 2,3,6-trideoxy sugar nucleotides were described for the first time. The study of their hydrolysis kinetics in aqueous buffers provided insight into their application as glycosyl donors.
Co-reporter:Pu Hua Wu, Qing Qing Meng, Hu Chen Zhou
Chinese Chemical Letters 2011 Volume 22(Issue 12) pp:1411-1414
Publication Date(Web):December 2011
DOI:10.1016/j.cclet.2011.06.005
A novel pyrrolo-benzoxaborole, 6-(pyrrol-1-yl)-1,3-dihydro-1-hydroxy-2,1-benzoxaborole, was synthesized with 27% overall yield over six steps from 2-bromo-1-methyl-4-nitrobenzene as starting material. Its derivatization was achieved via Friedel–Crafts reaction catalyzed by anhydrous stannic chloride with various acyl chlorides giving 3-acyl-1-phenylpyrroles as the main products.
Co-reporter:Zhitao Qiao, Weiwei Wang, Lie Wang, Donghua Wen, Yaxue Zhao, Qing Wang, Qingqing Meng, Guoqiang Chen, Yingli Wu, Huchen Zhou
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 21) pp:6389-6392
Publication Date(Web):1 November 2011
DOI:10.1016/j.bmcl.2011.08.101
As the best-characterized ubiquitin-like protein (UBL), small ubiquitin-related modifier (SUMO) was found to conjugate with a number of proteins to regulate cellular functions including transcription, signal transduction, and cell cycle. While E1, E2 and E3 ligases are responsible for the forward SUMOylation reaction, SUMO-specific proteases (SENPs) reversibly remove SUMO from the SUMOylated proteins. Recently, SENP1 was found to be a potential therapeutic target for the treatment of prostate cancers, but the design and synthesis of its inhibitors have not been reported. We designed and synthesized a series of benzodiazepine-based SENP1 inhibitors, and they showed inhibitory activity as good as IC50 = 9.2 μM (compound 38). The structure–activity relationship was also discussed.
Co-reporter:Dazhong Ding, Yaxue Zhao, Qingqing Meng, Dongsheng Xie, Bakela Nare, Daitao Chen, Cyrus J. Bacchi, Nigel Yarlett, Yong-Kang Zhang, Vincent Hernandez, Yi Xia, Yvonne Freund, Maha Abdulla, Kean-Hooi Ang, Joseline Ratnam, James H. McKerrow, Robert T. Jacobs, Huchen Zhou and Jacob J. Plattner
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 4) pp:165
Publication Date(Web):April 6, 2010
DOI:10.1021/ml100013s
We report the discovery of benzoxaborole antitrypanosomal agents and their structure−activity relationships on central linkage groups and different substitution patterns in the sulfur-linked series. The compounds showed in vitro growth inhibition IC50 values as low as 0.02 μg/mL and in vivo efficacy in acute murine infection models against Tryapnosoma brucei.Keywords (keywords): African trypanosomiasis; benzoxaborole; Tryapnosoma brucei
Co-reporter:Long Ye, Dazhong Ding, Yiqing Feng, Dongsheng Xie, Puhua Wu, Hui Guo, Qingqing Meng, Huchen Zhou
Tetrahedron 2009 65(42) pp: 8738-8744
Publication Date(Web):
DOI:10.1016/j.tet.2009.08.026
Co-reporter:Jiong Zhang, Fei Yang, Zhitao Qiao, Mingyan Zhu, Huchen Zhou
Bioorganic & Medicinal Chemistry Letters (1 December 2016) Volume 26(Issue 23) pp:
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.024
In this study, we report the synthesis of a series of chalcone–benzoxaborole hybrid molecules and the evaluation of their anticancer activity. Their anticancer potency and toxicity were tested on three human cancer cell lines and two normal cell lines. The 4-fluoro compound 15 was found to be the most potent compound with an IC50 value of 1.4 μM on SKOV3 cells. The 4-iodo compound 18 and 3-methyloxy-4-amino compound 47 showed good potency on SKOV3 cells while exhibiting low toxicity on normal cells. This work extended the application of benzoxaboroles to the field of anticancer research.
Co-reporter:Jiong Zhang, Fei Yang, Zhitao Qiao, Mingyan Zhu, Huchen Zhou
Bioorganic & Medicinal Chemistry Letters (1 December 2016) Volume 26(Issue 23) pp:5797-5801
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.024
In this study, we report the synthesis of a series of chalcone–benzoxaborole hybrid molecules and the evaluation of their anticancer activity. Their anticancer potency and toxicity were tested on three human cancer cell lines and two normal cell lines. The 4-fluoro compound 15 was found to be the most potent compound with an IC50 value of 1.4 μM on SKOV3 cells. The 4-iodo compound 18 and 3-methyloxy-4-amino compound 47 showed good potency on SKOV3 cells while exhibiting low toxicity on normal cells. This work extended the application of benzoxaboroles to the field of anticancer research.
Co-reporter:Fenglong Zhang, Jin Du, Qing Wang, Qinghua Hu, Jiong Zhang, Dazhong Ding, Yaxue Zhao, Fei Yang, Enduo Wang and Huchen Zhou
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 32) pp:NaN5324-5324
Publication Date(Web):2013/06/19
DOI:10.1039/C3OB40236C
Human African trypanosomiasis (HAT) is one of the most neglected diseases in the tropic regions, which is fatal if not treated in time. There is an urgent need for new therapeutics, especially those in new chemical classes. Leucyl-tRNA synthetase (LeuRS) has been paid much attention as a recently clinically validated antimicrobial target. Our group has previously reported T. brucei LeuRS (TbLeuRS) inhibitors, including benzoxaboroles targeting the editing site and pyrrolinones targeting the synthetic site. Here we report the discovery of N-(4-sulfamoylphenyl)thioureas as a new class of TbLeuRS inhibitors. The R1 and R2 groups, reminiscent of the leucyl and adenyl regions of aa-AMP and aa-AMS, were optimized to result in a significant 13-fold increase of inhibitory activity (compound 19, IC50 = 13.7 μM). Aided by ligand–protein docking, the 1,3-substitution at the central phenyl ring was predicted and proved to give significantly improved activity (59, IC50 = 1.1 μM). This work provided a new scaffold for the exploration of novel inhibitors against TbLeuRS, which may become potential therapeutics for the treatment of HAT.
Co-reporter:Yaxue Zhao, Feng Zhou, Huchen Zhou and Haibin Su
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 5) pp:NaN1698-1698
Publication Date(Web):2012/11/28
DOI:10.1039/C2CP42830J
The bonding characteristics in cysteine–gold cluster complexes represented by thiolate (Aun·CysS (n = 1, 3, 5, 7)) and thiol (Aun·CysSH (n = 2, 4, 6, 8)) is investigated by density functional theory with 6-31G(d,p) and Lanl2DZ hybrid basis sets. The complexes exhibit very different bonding characteristic between these two forms. In the Aun·CysS complexes, the charge transfers from gold clusters to sulfur atoms. The number of S–Au bonds in the Aun·CysS complexes evolves from one to two when n is greater than three. For n equals three, i.e. Au3·CysS, its ground state only has one S–Au bond. While the only S–Au bond in Au1·CysS is mainly covalent, the nature of the S–Au bond in other thiolates is featured with the combination of covalent and donor–acceptor interactions. In particular, one stable isomer of Au3·CysS with two S–Au bonds, which is 2 kcal mol−1 higher in energy than the corresponding ground state, consists of one covalent and one donor–acceptor S–Au bond explicitly. Moreover, the localized three center two electron bonds are formed within the Au clusters, which facilitates the formation of the two S–Au bonds in Au5·CysS and Au7·CysS complexes. In the Aun·CysSH complexes, the donor–acceptor interaction prevails in the Au–SH bond by transferring lone pair electrons from the sulfur atom to the adjacent gold atom. Interestingly, the orbital with much more 6s-component in Au4·CysSH enhances the donor–acceptor bonding character, thus yields the strongest bonding among all the Aun·CysSH complexes studied in this paper. In general, the bonding strength between gold clusters and cysteine is positively correlated with the S–Au overlap-weighted bond order, but negatively correlated with the S–Au bond length. Lastly, the covalent and donor–acceptor S–Au bond strength is computed to be 48 and 18 kcal mol−1, respectively.

![<br>1-(4-chlorophenyl)-2-oxo-2-phenylethyl ({[(4-bromobenzoyl)amino]acetyl}amin o)acetate](http://img.cochemist.com/ccimg/665000/664976-49-2.png)
![<br>1-(4-chlorophenyl)-2-oxo-2-phenylethyl ({[(4-bromobenzoyl)amino]acetyl}amin o)acetate](http://img.cochemist.com/ccimg/665000/664976-49-2_b.png)
![Ethanone, 1-[3-(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/124500/124414-06-8.png)
![Ethanone, 1-[3-(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/124500/124414-06-8_b.png)
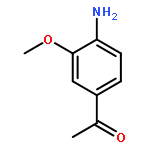
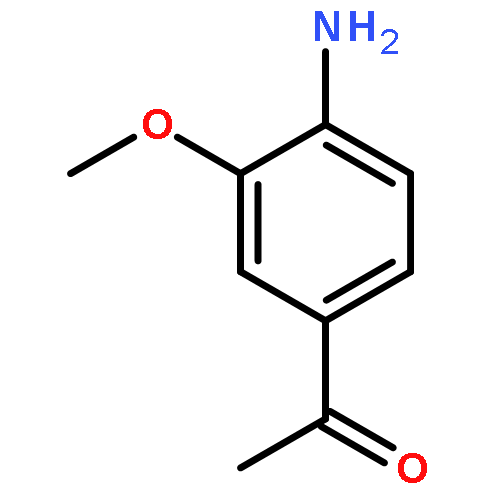
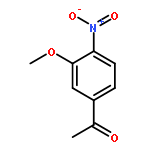
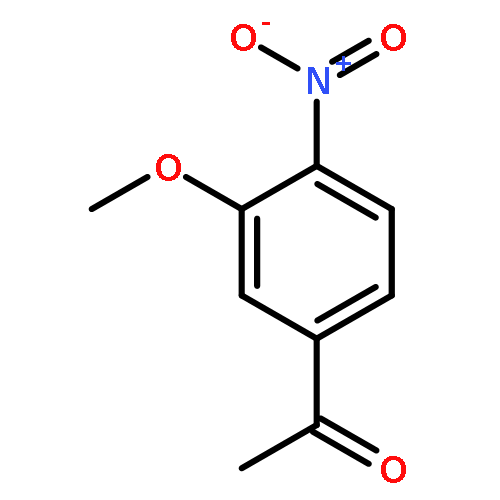
![<br>3-nitro-N-(3-{[(2E)-2-(3-nitrobenzylidene)hydrazino]carbonyl}phenyl)benzami de](http://img.cochemist.com/ccimg/405100/405090-87-1.png)
![<br>3-nitro-N-(3-{[(2E)-2-(3-nitrobenzylidene)hydrazino]carbonyl}phenyl)benzami de](http://img.cochemist.com/ccimg/405100/405090-87-1_b.png)