Co-reporter:Ahmed E. Fouda ;Mary Kay H. Pflum
Angewandte Chemie International Edition 2015 Volume 54( Issue 33) pp:9618-9621
Publication Date(Web):
DOI:10.1002/anie.201503041
Abstract
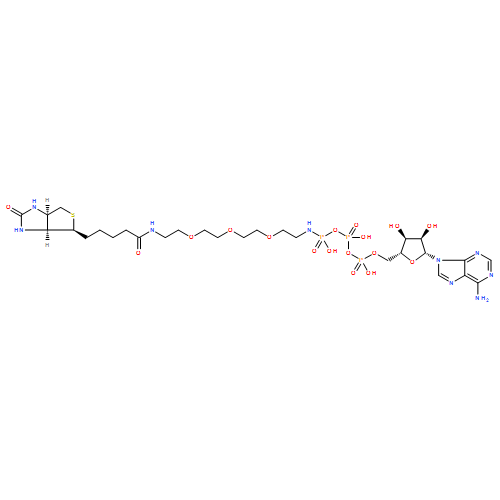
ATP analogues have been powerful compounds for the study of kinase-catalyzed phosphorylation. However, the cell impermeability of ATP analogues has largely limited their use to in vitro lysate-based experiments. Herein, we report the first cell-permeable ATP analogue, ATP–polyamine–biotin (APB). APB is shown to promote biotin labeling of kinase substrates in live cells and has future applications in phosphoprotein purification and analysis. More generally, these studies provide a foundation for the development of additional cell-permeable ATP analogues for cell-signaling research.
Co-reporter:Ahmed E. Fouda ;Mary Kay H. Pflum
Angewandte Chemie 2015 Volume 127( Issue 33) pp:9754-9757
Publication Date(Web):
DOI:10.1002/ange.201503041
Abstract
ATP analogues have been powerful compounds for the study of kinase-catalyzed phosphorylation. However, the cell impermeability of ATP analogues has largely limited their use to in vitro lysate-based experiments. Herein, we report the first cell-permeable ATP analogue, ATP–polyamine–biotin (APB). APB is shown to promote biotin labeling of kinase substrates in live cells and has future applications in phosphoprotein purification and analysis. More generally, these studies provide a foundation for the development of additional cell-permeable ATP analogues for cell-signaling research.
Co-reporter:Chamara Senevirathne ; Mary Kay H. Pflum
ChemBioChem 2013 Volume 14( Issue 3) pp:381-387
Publication Date(Web):
DOI:10.1002/cbic.201200626
Abstract
Kinase-catalyzed protein phosphorylation is involved in a wide variety of cellular events. Development of methods to monitor phosphorylation is critical to understand cell biology. Our lab recently discovered kinase-catalyzed biotinylation, where ATP-biotin is utilized by kinases to label phosphopeptides or phosphoproteins with a biotin tag. To exploit kinase-catalyzed biotinylation for phosphoprotein purification and identification in a cellular context, the susceptibility of the biotin tag to phosphatases was characterized. We found that the phosphorylbiotin group on peptide and protein substrates was relatively insensitive to protein phosphatases. To understand how phosphatase stability would impact phosphoproteomics research applications, kinase-catalyzed biotinylation of cell lysates was performed in the presence of kinase or phosphatase inhibitors. We found that biotinylation with ATP-biotin was sensitive to inhibitors, although with variable effects compared to ATP phosphorylation. The results suggest that kinase-catalyzed biotinylation is well suited for phosphoproteomics studies, with particular utility towards monitoring low-abundance phosphoproteins or characterizing the influence of inhibitor drugs on protein phosphorylation.
Co-reporter:Sujit Suwal, Chamara Senevirathne, Satish Garre, and Mary Kay H. Pflum
Bioconjugate Chemistry 2012 Volume 23(Issue 12) pp:2386
Publication Date(Web):November 1, 2012
DOI:10.1021/bc300404s
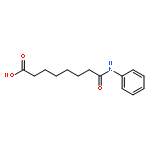
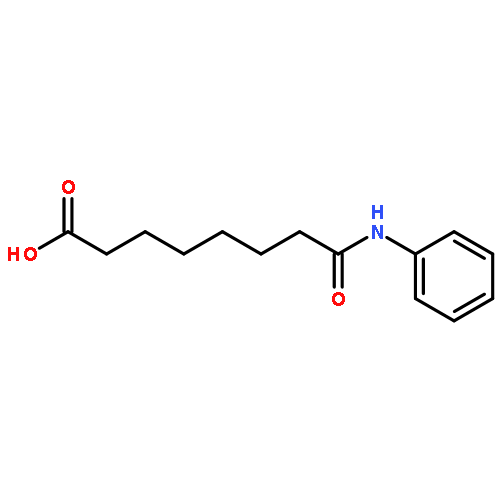
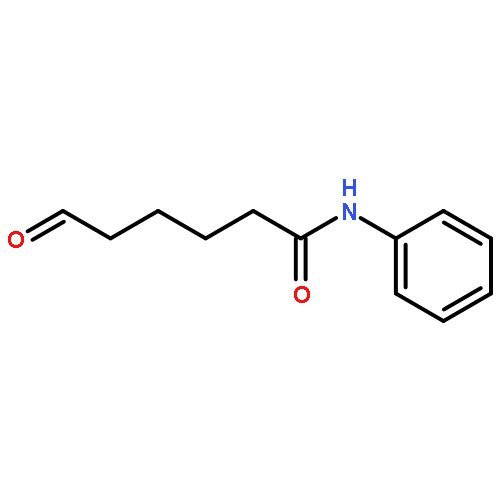
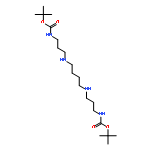
Kinase-catalyzed protein phosphorylation is an important biochemical process involved in cellular functions. We recently discovered that kinases promiscuously accept γ-modified ATP analogues as cosubstrates and used several ATP analogues as tools for studying protein phosphorylation. Herein, we explore the structural requirements of γ-modified ATP analogues for kinase compatibility. To understand the influence of linker length and composition, a series of ATP analogues was synthesized, and the efficiency of kinase-catalyzed labeling was determined by quantitative mass spectrometry. This study on factors influencing kinase cosubstrate promiscuity will enable design of ATP analogues for a variety of kinase-catalyzed labeling reactions.
Co-reporter:Sun Ea Choi, Mary Kay H. Pflum
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 23) pp:7084-7086
Publication Date(Web):1 December 2012
DOI:10.1016/j.bmcl.2012.09.093
Suberoylanilide hydroxamic acid (SAHA, Vorinostat), the first FDA-approved histone deacetylase (HDAC) inhibitor drug, was modified at the C6 position to study the structural requirements for high potency and selectivity. Substituents on the C6 position only modestly influenced inhibitor potency, with poorer activity observed as substituent size increased. Interestingly, C6 substituents also modestly influenced selectivity compared to the parent compound, SAHA. This systematic study documenting the influence of substituents on the SAHA linker region will aid development of anti-cancer drugs targeting HDAC proteins.
Co-reporter:Sun Ea Choi, Sujith V.W. Weerasinghe, Mary Kay H. Pflum
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 20) pp:6139-6142
Publication Date(Web):15 October 2011
DOI:10.1016/j.bmcl.2011.08.027
The FDA-approved drug suberoylanilide hydroxamic acid (SAHA, Vorinostat) was modified to improve its selectivity for a single histone deaetylase (HDAC) isoform. We show that attaching an ethyl group at the C3 position transforms SAHA from nonselective to an HDAC6-selective inhibitor. Theses results indicate that small structural changes in SAHA can significantly influence selectivity, which will lead future anti-cancer design efforts targeting HDAC proteins.
Co-reporter:Sujith V.W. Weerasinghe, Magdalene Wambua, Mary Kay H. Pflum
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 21) pp:7586-7592
Publication Date(Web):1 November 2010
DOI:10.1016/j.bmc.2010.08.045
Histone deacetylase (HDAC) proteins are promising targets for cancer treatment, as shown by the recent FDA approval of the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA, Vorinostat) for the treatment of cutaneous T-cell lymphoma. To identify additional potent inhibitors and characterize HDAC mutant proteins, there is interest to develop an inexpensive screening method dependent on native substrates. Here, we report the first yeast-based gene reporter screen dependent on the yeast Rpd3, which is a homolog of human class I HDAC proteins. The screen was sensitive to an inactive Rpd3 mutant and various inhibitors in qualitative, agar-based and quantitative, solution-phase formats. Interestingly, inclusion of the lytic enzyme zymolyase enhanced the inhibitor sensitivity of the screen. The gene reporter screen provides a tool to screen Rpd3 mutants and inhibitors of class I HDAC proteins.
Co-reporter:Sujit Suwal ;MaryKayH. Pflum
Angewandte Chemie International Edition 2010 Volume 49( Issue 9) pp:1627-1630
Publication Date(Web):
DOI:10.1002/anie.200905244
Co-reporter:Sujit Suwal ;MaryKayH. Pflum
Angewandte Chemie 2010 Volume 122( Issue 9) pp:1671-1674
Publication Date(Web):
DOI:10.1002/ange.200905244
Co-reporter:Keith D. Green Dr.;Mary Kay H. Pflum
ChemBioChem 2009 Volume 10( Issue 2) pp:234-237
Publication Date(Web):
DOI:10.1002/cbic.200800393
Co-reporter:Anton V. Bieliauskas and Mary Kay H. Pflum
Chemical Society Reviews 2008 vol. 37(Issue 7) pp:1402-1413
Publication Date(Web):08 May 2008
DOI:10.1039/B703830P
Histone deacetylase (HDAC) proteins are transcription regulators linked to cancer. As a result, multiple small molecule HDAC inhibitors are in various phases of clinical trials as anti-cancer drugs. The majority of HDAC inhibitors non-selectively influence the activities of eleven human HDAC isoforms, which are divided into distinct classes. This tutorial review focuses on the recent progress toward the identification of class-selective and isoform-selective HDAC inhibitors. The emerging trends suggest that subtle differences in the active sites of the HDAC isoforms can be exploited to dictate selectivity.
Co-reporter:Sujith V. W. Weerasinghe ; Guillermina Estiu ; Olaf Wiest ;Mary Kay H. Pflum
Journal of Medicinal Chemistry 2008 Volume 51(Issue 18) pp:5542-5551
Publication Date(Web):August 27, 2008
DOI:10.1021/jm800081j
Histone deacetylase 1 (HDAC1) has been linked to cell growth and cell cycle regulation, which makes it a widely recognized target for anticancer drugs. Whereas variations of the metal-binding and capping groups of HDAC inhibitors have been studied extensively, the role of the linker region is less well known, despite the potency of inhibitors with diverse linkers, such as MS-275. To facilitate a drug design that targets HDAC1, we assessed the influence of residues in the 11 Å channel of the HDAC1 active site on activity by using an alanine scan. The mutation of eight channel residues to alanine resulted in a substantial reduction in deacetylase activity. Molecular dynamics simulations indicated that alanine mutation results in significant movement of the active-site channel, which suggests that channel residues promote HDAC1 activity by influencing substrate interactions. With little characterization of HDAC1 available, the combined experimental and computational results define the active-site residues of HDAC1 that are critical for substrate/inhibitor binding and provide important insight into drug design.
Co-reporter:Anton V. Bieliauskas and Mary Kay H. Pflum
Chemical Society Reviews 2008 - vol. 37(Issue 7) pp:NaN1413-1413
Publication Date(Web):2008/05/08
DOI:10.1039/B703830P
Histone deacetylase (HDAC) proteins are transcription regulators linked to cancer. As a result, multiple small molecule HDAC inhibitors are in various phases of clinical trials as anti-cancer drugs. The majority of HDAC inhibitors non-selectively influence the activities of eleven human HDAC isoforms, which are divided into distinct classes. This tutorial review focuses on the recent progress toward the identification of class-selective and isoform-selective HDAC inhibitors. The emerging trends suggest that subtle differences in the active sites of the HDAC isoforms can be exploited to dictate selectivity.









![Acetamide,N-[2-[2-(2-aminoethoxy)ethoxy]ethyl]-](http://img.cochemist.com/ccimg/866500/866404-69-5.png)
![Acetamide,N-[2-[2-(2-aminoethoxy)ethoxy]ethyl]-](http://img.cochemist.com/ccimg/866500/866404-69-5_b.png)

![Benzamide, 4-(acetylamino)-N-[2-amino-5-(2-thienyl)phenyl]-](http://img.cochemist.com/ccimg/849300/849234-64-6.png)
![Benzamide, 4-(acetylamino)-N-[2-amino-5-(2-thienyl)phenyl]-](http://img.cochemist.com/ccimg/849300/849234-64-6_b.png)































![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7.png)
![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7_b.png)







![5'-O-[{[{[(benzylamino)(hydroxy)phosphoryl]oxy}(hydroxy)phosphoryl]oxy}(hydroxy)phosphoryl]adenosine](http://img.cochemist.com/ccimg/58100/58026-10-1.png)
![5'-O-[{[{[(benzylamino)(hydroxy)phosphoryl]oxy}(hydroxy)phosphoryl]oxy}(hydroxy)phosphoryl]adenosine](http://img.cochemist.com/ccimg/58100/58026-10-1_b.png)
![5'-O-{hydroxy[(hydroxy{[hydroxy(phenylamino)phosphoryl]oxy}phosphoryl)oxy]phosphoryl}adenosine](http://img.cochemist.com/ccimg/52900/52830-41-8.png)
![5'-O-{hydroxy[(hydroxy{[hydroxy(phenylamino)phosphoryl]oxy}phosphoryl)oxy]phosphoryl}adenosine](http://img.cochemist.com/ccimg/52900/52830-41-8_b.png)