Co-reporter:Isabelle I. Salles, Hendrik B. Feys, Brecht F. Iserbyt, Simon F. De Meyer, Karen Vanhoorelbeke, Hans Deckmyn
Blood Reviews (May 2008) Volume 22(Issue 3) pp:155-172
Publication Date(Web):1 May 2008
DOI:10.1016/j.blre.2007.11.002
Inherited platelet disorders constitute a large group of diseases involving a wide range of genetic defects that can lead to bleeding symptoms of varying severity. They are associated with defects in surface membrane glycoproteins resulting in e.g. Bernard Soulier Syndrome and Glanzmann Thrombasthenia causing defects in platelet adhesion and aggregation, respectively, as well as in receptors for agonists (a.o. P2Y12, TXA2) disrupting platelet signalling. Defects affecting platelet granules can be characterised by abnormalities of α-granules as in the Gray platelet syndrome or dense granules as in Hermansky-Pudlak and Chediak-Higashi syndromes, the latter two also altering other cytoplasmic organelles such as melanosomes and therefore not restricted to platelets. Finally, defects in proteins essential to signalling pathways (a.o. in Wiskott-Aldrich syndrome) or in platelet-derived procoagulant activity (Scott and Stormorken syndromes) also impair platelet function. For most of the above disorders mouse knockout models have been generated, that allowed to confirm the genotype-phenotype relationship and to further unravel the molecular causes of the disease and the mechanisms underlying primary haemostasis. More recently, interest has been growing in the effects of the more common polymorphisms that are found in the platelet glycoproteins as possible risk factors for thrombotic disorders. The new era of platelet genomics and proteomics will increase our knowledge on platelet disorders that will improve their diagnosis, but also will provide basis for new antithrombotic therapies.
Co-reporter:Katleen Broos, Hendrik B. Feys, Simon F. De Meyer, Karen Vanhoorelbeke, Hans Deckmyn
Blood Reviews (July 2011) Volume 25(Issue 4) pp:
Publication Date(Web):1 July 2011
DOI:10.1016/j.blre.2011.03.002
When platelet numbers are low or when their function is disabled, the risk of bleeding is high, which on the one hand indicates that in normal life vascular damage is a rather common event and that hence the role of platelets in maintaining a normal hemostasis is a continuously ongoing physiological process. Upon vascular injury, platelets instantly adhere to the exposed extracellular matrix resulting in platelet activation and aggregation to form a hemostatic plug. This self-amplifying mechanism nevertheless requires a tight control to prevent uncontrolled platelet aggregate formation that eventually would occlude the vessel. Therefore endothelial cells produce inhibitory compounds such as prostacyclin and nitric oxide that limit the growth of the platelet thrombus to the damaged area. With this review, we intend to give an integrated survey of the platelet response to vascular injury in normal hemostasis.

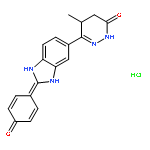
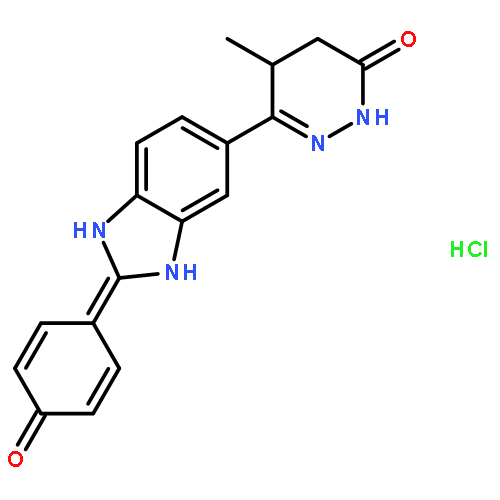
![3(2H)-Pyridazinone, 4,5-dihydro-6-[2-(4-hydroxyphenyl)-1H-benzimidazol-5-yl]-5-methyl-](http://img.cochemist.com/ccimg/108400/108381-22-2.png)
![3(2H)-Pyridazinone, 4,5-dihydro-6-[2-(4-hydroxyphenyl)-1H-benzimidazol-5-yl]-5-methyl-](http://img.cochemist.com/ccimg/108400/108381-22-2_b.png)
![7-[3-(3-HYDROXYOCT-1-ENYL)-4,6-DIOXABICYCLO[3.1.1]HEPT-2-YL]HEPT-5-ENOIC ACID](http://img.cochemist.com/ccimg/57600/57576-52-0.png)
![7-[3-(3-HYDROXYOCT-1-ENYL)-4,6-DIOXABICYCLO[3.1.1]HEPT-2-YL]HEPT-5-ENOIC ACID](http://img.cochemist.com/ccimg/57600/57576-52-0_b.png)


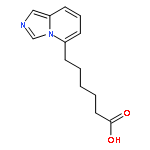
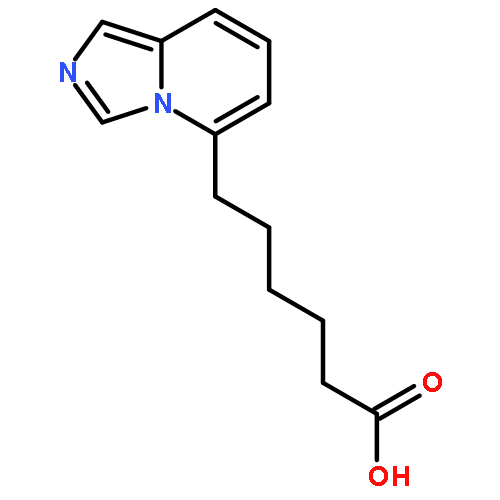
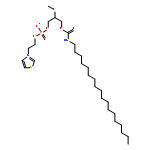
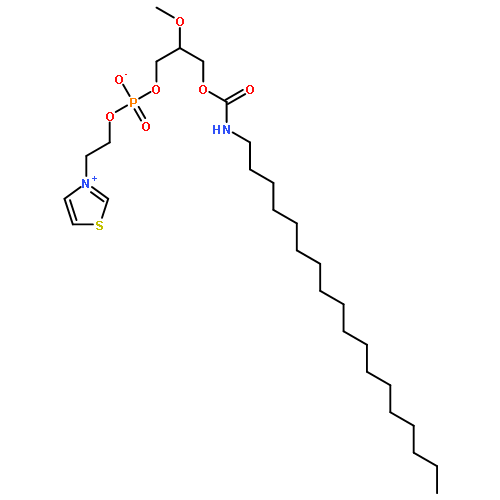
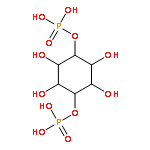
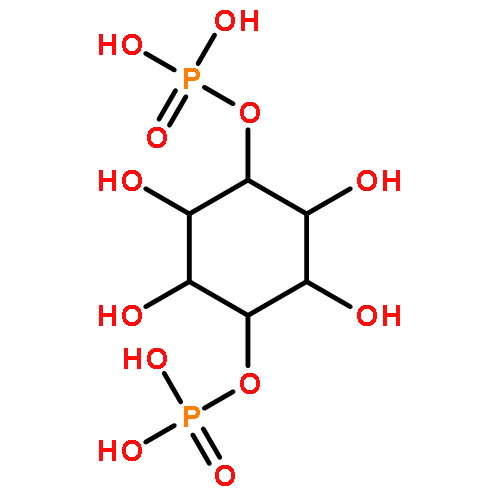
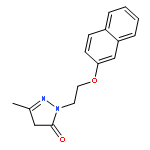
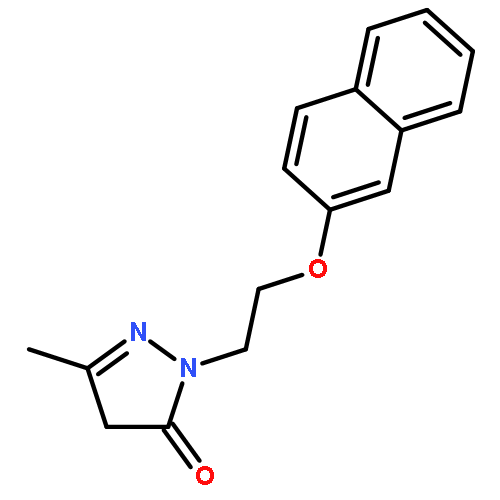




![4-Benzoxazolecarboxylicacid,5-(methylamino)-2-[[(2R,3R,6S,8S,9R,11R)-3,9,11-trimethyl-8-[(1S)-1-methyl-2-oxo-2-(1H-pyrrol-2-yl)ethyl]-1,7-dioxaspiro[5.5]undec-2-yl]methyl]-](http://img.cochemist.com/ccimg/52700/52665-69-7.png)
![4-Benzoxazolecarboxylicacid,5-(methylamino)-2-[[(2R,3R,6S,8S,9R,11R)-3,9,11-trimethyl-8-[(1S)-1-methyl-2-oxo-2-(1H-pyrrol-2-yl)ethyl]-1,7-dioxaspiro[5.5]undec-2-yl]methyl]-](http://img.cochemist.com/ccimg/52700/52665-69-7_b.png)




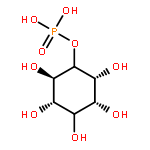
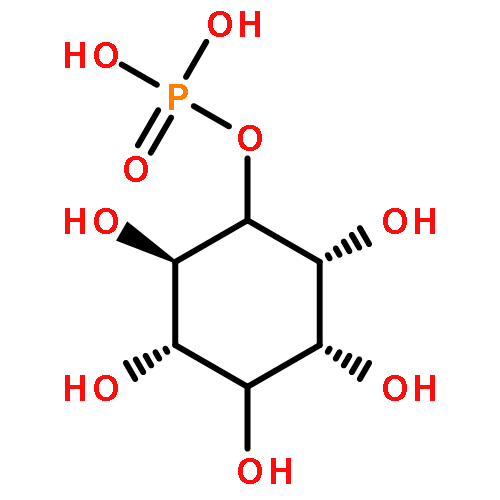
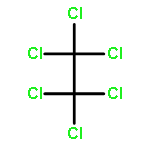
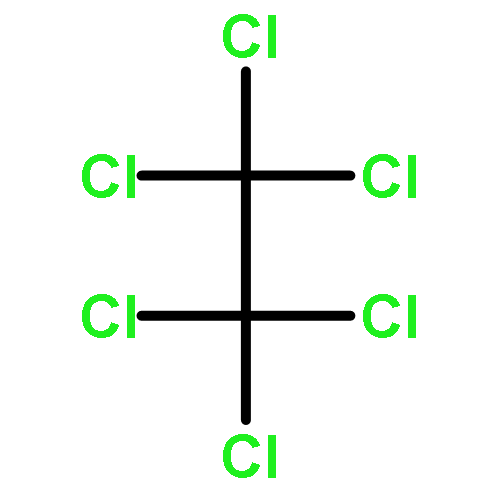
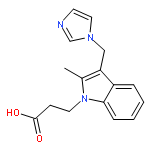
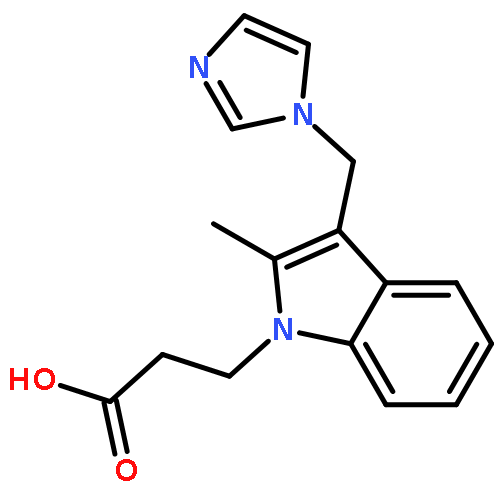
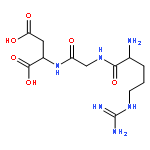

![[(3-FLUOROBENZYL)OXY]AMMONIUM CHLORIDE](http://img.cochemist.com/ccimg/72200/72131-33-0.png)
![[(3-FLUOROBENZYL)OXY]AMMONIUM CHLORIDE](http://img.cochemist.com/ccimg/72200/72131-33-0_b.png)
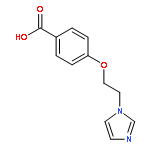
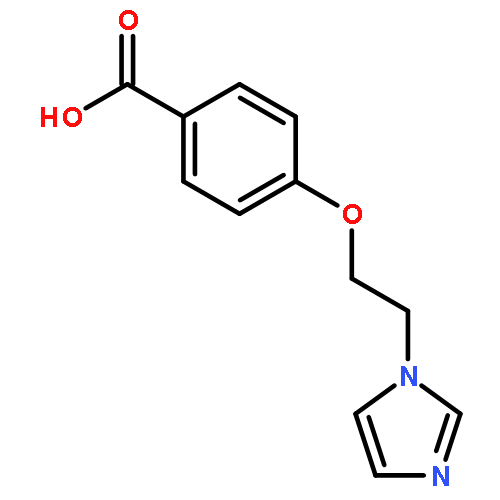
![(E)-2-METHYL-3-[4-(PYRIDIN-3-YLMETHYL)PHENYL]PROP-2-ENOIC ACID](http://img.cochemist.com/ccimg/76000/75987-08-5.png)
![(E)-2-METHYL-3-[4-(PYRIDIN-3-YLMETHYL)PHENYL]PROP-2-ENOIC ACID](http://img.cochemist.com/ccimg/76000/75987-08-5_b.png)
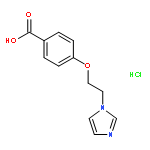
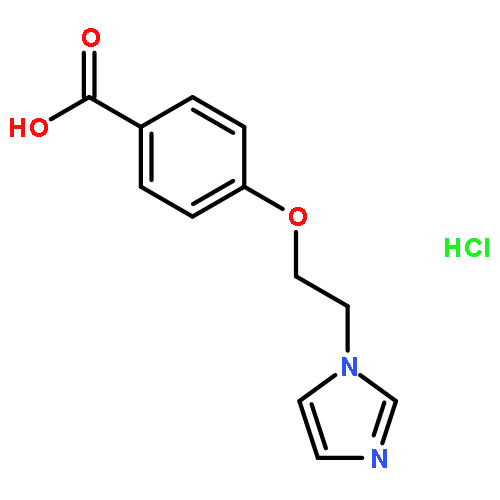
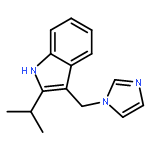
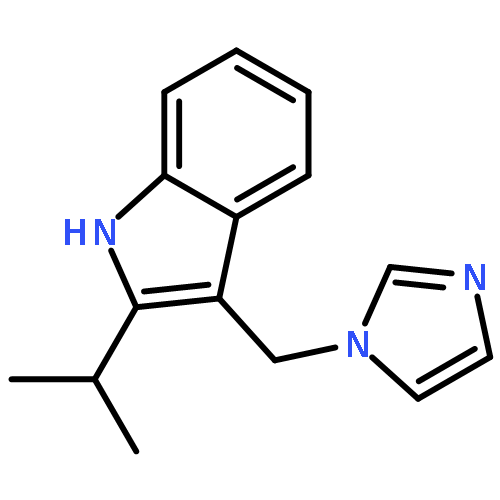






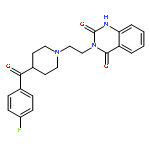
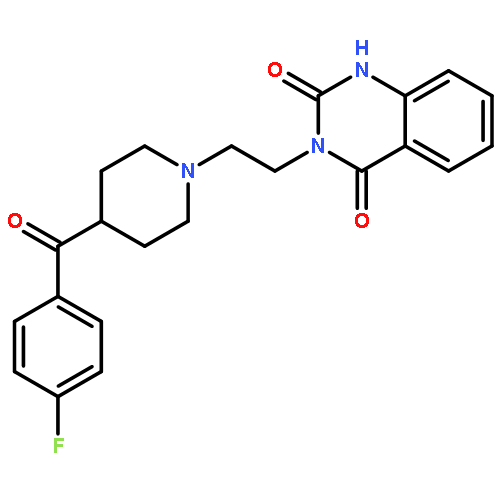
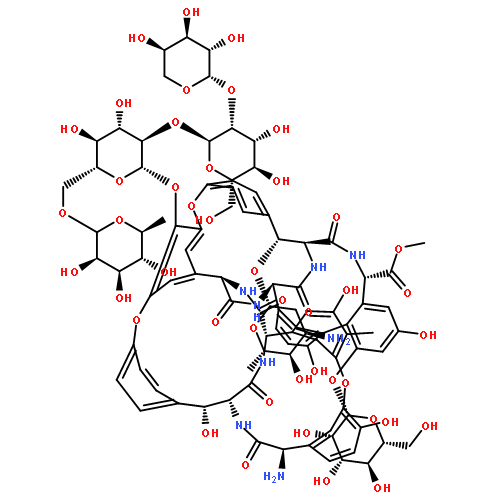


![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2.png)
![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2_b.png)
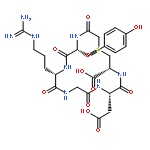
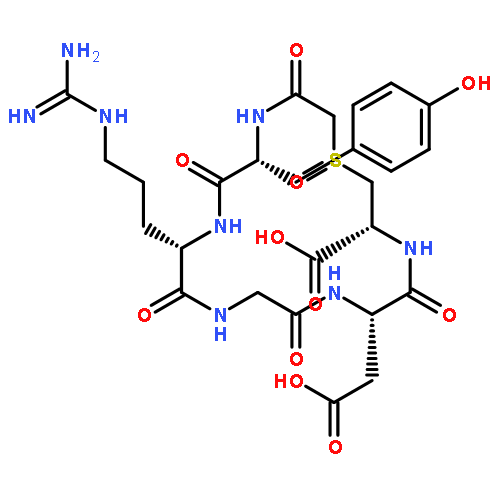

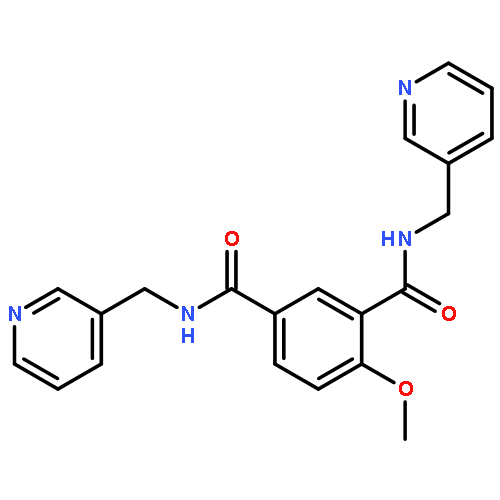


![Benzo[b]thiophene-2-carboxamide,3-hydroxy-N-[2-phenyl-2-(2-thienyl)ethenyl]-5-(trifluoromethyl)-, (E)- (9CI)](http://img.cochemist.com/ccimg/102600/102565-09-3.png)
![Benzo[b]thiophene-2-carboxamide,3-hydroxy-N-[2-phenyl-2-(2-thienyl)ethenyl]-5-(trifluoromethyl)-, (E)- (9CI)](http://img.cochemist.com/ccimg/102600/102565-09-3_b.png)
![Benzo[b]thiophene-2-carboxamide,3-hydroxy-N-[2-phenyl-2-(2-thienyl)ethenyl]-5-(trifluoromethyl)-](http://img.cochemist.com/ccimg/107100/107008-29-7.png)
![Benzo[b]thiophene-2-carboxamide,3-hydroxy-N-[2-phenyl-2-(2-thienyl)ethenyl]-5-(trifluoromethyl)-](http://img.cochemist.com/ccimg/107100/107008-29-7_b.png)
![Pentanoic acid,5-[[(E)-[3-pyridinyl[3-(trifluoromethyl)phenyl]methylene]amino]oxy]-](http://img.cochemist.com/ccimg/110200/110140-89-1.png)
![Pentanoic acid,5-[[(E)-[3-pyridinyl[3-(trifluoromethyl)phenyl]methylene]amino]oxy]-](http://img.cochemist.com/ccimg/110200/110140-89-1_b.png)










![5-Heptenoic acid,7-[(1S,4R,5S,6R)-5-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2-oxabicyclo[2.2.1]hept-6-yl]-,(5Z)-](http://img.cochemist.com/ccimg/57000/56985-32-1.png)
![5-Heptenoic acid,7-[(1S,4R,5S,6R)-5-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2-oxabicyclo[2.2.1]hept-6-yl]-,(5Z)-](http://img.cochemist.com/ccimg/57000/56985-32-1_b.png)