Co-reporter:Lauren Seabrooks, Longqin Hu
Acta Pharmaceutica Sinica B 2017 Volume 7, Issue 4(Issue 4) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.apsb.2017.05.001
Nature has been the source of life-changing and -saving medications for centuries. Aspirin, penicillin and morphine are prime examples of Nature׳s gifts to medicine. These discoveries catalyzed the field of natural product drug discovery which has mostly focused on plants. However, insects have more than twice the number of species and entomotherapy has been in practice for as long as and often in conjunction with medicinal plants and is an important alternative to modern medicine in many parts of the world. Herein, an overview of current traditional medicinal applications of insects and characterization of isolated biologically active molecules starting from approximately 2010 is presented. Insect natural products reviewed were isolated from ants, bees, wasps, beetles, cockroaches, termites, flies, true bugs, moths and more. Biological activities of these natural products from insects include antimicrobial, antifungal, antiviral, anticancer, antioxidant, anti-inflammatory and immunomodulatory effects.This review presents an overview of current traditional medicinal applications of insects and characterization of isolated biologically active molecules starting from approximately 2010.Download high-res image (220KB)Download full-size image
Co-reporter:Longqin Hu, Yanhui Yang, Herve Aloysius, Haifa Albanyan, Min Yang, Jian-Jie Liang, Anthony Yu, Alexander Shtukenberg, Laura N. Poloni, Vladyslav Kholodovych, Jay A. Tischfield, David S. Goldfarb, Michael D. Ward, and Amrik Sahota
Journal of Medicinal Chemistry 2016 Volume 59(Issue 15) pp:7293-7298
Publication Date(Web):July 13, 2016
DOI:10.1021/acs.jmedchem.6b00647
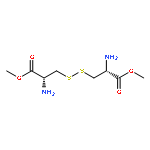
l-Cystine bismorpholide (1a) and l-cystine bis(N′-methylpiperazide) (1b) were seven and twenty-four times more effective than l-cystine dimethyl ester (CDME) in increasing the metastable supersaturation range of l-cystine, respectively, effectively inhibiting l-cystine crystallization. This behavior can be attributed to inhibition of crystal growth at microscopic length scale, as revealed by atomic force microscopy. Both 1a and 1b are more stable than CDME, and 1b was effective in vivo in a knockout mouse model of cystinuria.
Co-reporter:Xinghua Wu, Longqin Hu
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 12) pp:2697-2706
Publication Date(Web):15 June 2016
DOI:10.1016/j.bmc.2016.04.035
A series of Glutaryl-Hyp-Ala-Ser-Chg-Gln-4-aminobenzyl phosphoramide mustard conjugates (1a–e) was designed and synthesized as potential prodrugs for site-specific activation by PSA in prostate cancer cells. All conjugates were found to be substrates of PSA with cleavage occurring between Gln and the para-aminobenzyl (PAB) linker. Structure–activity relationship studies on these conjugates indicated that introduction of electron-withdrawing fluorine(s) on the phenyl ring in the PAB linker uniformly improved the chemical stability of the conjugates while the position of substitution affected differently the self-immolative process of conjugates upon proteolysis. Introduction of a fluorine at ortho position to benzylic phosphoramide as in 1b results in better stability of the conjugate prior to activation while maintaining its antiproliferative activity upon activation by PSA. The conjugate 1b with 2-fluoro substitution was identified as a promising lead for further evaluation and optimization in the development of prostate cancer-targeted prodrugs.
Co-reporter:Herve Aloysius
Chemical Biology & Drug Design 2015 Volume 86( Issue 4) pp:837-848
Publication Date(Web):
DOI:10.1111/cbdd.12559
To develop PSA peptide substrates with improved specificity and plasma stability from the known substrate sequence glutaryl-Hyp-Ala-Ser-Chg-Gln, systematic replacements of the N-terminal segment with D-retro-inverso-peptides were performed with the incorporation of 7-amino-4-methylcoumarin (7-AMC) after Gln for convenient fluorometric determination and ranking of the PSA substrate activity. The D-retro-inverso-peptide conjugates with P2-P5 D-amino acid substitutions were moderate but poorer PSA substrates as compared to the original peptide, suggesting that inversion of the amide bonds and/or incorporation of the additional atom as in the urea linker adversely affected PSA binding. However, P5 substitution of Hyp with Ser showed significant improvements in PSA cleavage rate; the resulting AMC conjugate, glutaryl-Ser-Ala-Ser-Chg-Gln-AMC (11), exhibited the fastest PSA cleavage rate of 351 pmol/min/100 nmol PSA. In addition, GABAmGly-Ala-Ser-Chg-Gln-AMC (conjugate 6) was the second best PSA substrate and released 7-AMC at a rate of 225 pmol/min/100 nmol PSA as compared to 171 pmol/min/100 nmol PSA for the control conjugate glutaryl-Hyp-Ala-Ser-Chg-Gln-AMC. Incubations of selected AMC conjugates with mouse and human plasma revealed that GABAD-Ser-ψ[NH-CO-NH]-Ala-Ser-Chg-Gln-AMC (5) and GABAmGly-Ala-Ser-Chg-Gln-AMC (6) were most stable to non-PSA-mediated proteolysis. Our results suggest that the PSA specificity of glutaryl-Hyp-Ala-Ser-Chg-Gln is improved with Ser and mGly substitutions of Hyp at the P5.
Co-reporter:Yongying Jiang, Longqin Hu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 23) pp:7507-7514
Publication Date(Web):1 December 2013
DOI:10.1016/j.bmc.2013.09.039
In our continued effort to develop prodrugs of phosphoramide mustard, conjugates of 4-aminocyclophosphamide (4-NH2-CPA) with three PSA-specific peptides were synthesized and evaluated as substrates of PSA. These include conjugates of cis-(2R,4R)-4-NH2-CPA with a tetrapeptide Succinyl-Ser-Lys-Leu-Gln-OH, a hexapeptide Succinyl-His-Ser-Ser-Lys-Leu-Gln-OH, and a pentapeptide Glutaryl-Hyp-Ala-Ser-Chg-Gln-OH. These conjugates were cleaved by PSA efficiently and exclusively after the expected glutamine residue to release 4-NH2-CPA, the activated prodrug form of phosphoramide mustard. The cleavage was most efficient for the pentapeptide conjugate 3 (Glutaryl-Hyp-Ala-Ser-Chg-Gln-NH-CPA), which showed a half-life of 55 min with PSA, followed by the hexapeptide conjugate 2 (Succinyl-His-Ser-Ser-Lys-Leu-Gln-NH-CPA) and the tertrapeptide conjugate 1 (Succinyl-Ser-Lys-Leu-Gln-NH-CPA) with half-lives of 6.5 and 12 h, respectively. These results indicate a potential of the conjugate 3 as an anticancer prodrug of phosphoramide mustard for selective PSA activation.
Co-reporter:Longqin Hu, Sadagopan Magesh, Lin Chen, Lili Wang, Timothy A. Lewis, Yu Chen, Carol Khodier, Daigo Inoyama, Lesa J. Beamer, Thomas J. Emge, Jian Shen, John E. Kerrigan, Ah-Ng Tony Kong, Sivaraman Dandapani, Michelle Palmer, Stuart L. Schreiber, Benito Munoz
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 10) pp:3039-3043
Publication Date(Web):15 May 2013
DOI:10.1016/j.bmcl.2013.03.013
A high-throughput screen (HTS) of the MLPCN library using a homogenous fluorescence polarization assay identified a small molecule as a first-in-class direct inhibitor of Keap1–Nrf2 protein–protein interaction. The HTS hit has three chiral centers; a combination of flash and chiral chromatographic separation demonstrated that Keap1-binding activity resides predominantly in one stereoisomer (SRS)-5 designated as ML334 (LH601A), which is at least 100× more potent than the other stereoisomers. The stereochemistry of the four cis isomers was assigned using X-ray crystallography and confirmed using stereospecific synthesis. (SRS)-5 is functionally active in both an ARE gene reporter assay and an Nrf2 nuclear translocation assay. The stereospecific nature of binding between (SRS)-5 and Keap1 as well as the preliminary but tractable structure–activity relationships support its use as a lead for our ongoing optimization
Co-reporter:Yanhui Yang, Andrew Voak, Shane R. Wilkinson, Longqin Hu
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 21) pp:6583-6586
Publication Date(Web):1 November 2012
DOI:10.1016/j.bmcl.2012.09.005
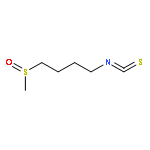
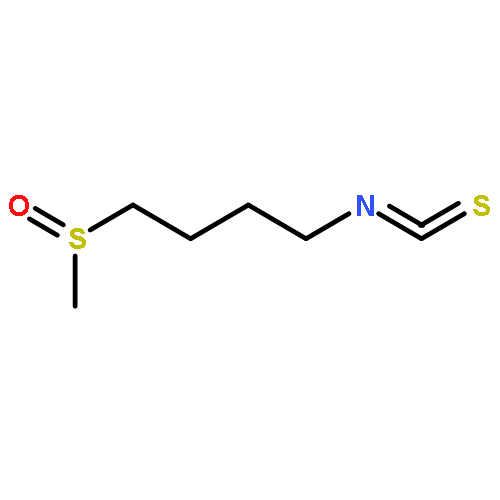
A series of potential DFMO prodrugs was designed through the incorporation of 4-nitrobenzyl ester or carbamate groups for potential activation by trypanosomal nitroreductase. It was found that only modification of Nε-amino group of DFMO by 4-nitro-2-fluorobenzyloxycarbonyl resulted in significant trypanocidal activity and could serve as a lead for further investigation.
Co-reporter:Yu Chen;Daigo Inoyama;Ah-Ng Tony Kong;Lesa J. Beamer
Chemical Biology & Drug Design 2011 Volume 78( Issue 6) pp:1014-1021
Publication Date(Web):
DOI:10.1111/j.1747-0285.2011.01240.x
The Keap1–Nrf2 interaction plays important roles in regulation of Nrf2 activity and induction of chemopreventive enzymes. To better understand the interaction and to determine the minimal Nrf2 sequence required for Keap1 binding, we synthesized a series of Nrf2 peptides containing ETGE motif and determined their binding affinities to the Kelch domain of Keap1 in solution using a surface plasmon resonance-based competition assay. The equilibrium dissociation constant for the interaction between 16mer Nrf2 peptide and Keap1 Kelch domain in solution () was found to be 23.9 nm, which is 10× lower than the surface binding constant () of 252 nm obtained for the direct binding of Keap1 Kelch domain to the immobilized 16mer Nrf2 peptide on a surface plasmon resonance sensor chip surface. The binding affinity of Nrf2 peptides to Keap1 Kelch domain was not lost until after deletion of eight residues from the N-terminus of the 16mer Nrf2 peptide. The 9mer Nrf2 peptide has a moderate binding affinity with a of 352 nm and the affinity was increased 15× upon removal of the positive charge at the peptide N-terminus by acetylation. These results suggest that the minimal Nrf2 peptide sequence required for Keap1 binding is the 9mer sequence of LDEETGEFL.
Co-reporter:Longqin Hu, Xinghua Wu, Jiye Han, Lin Chen, Simon O. Vass, Patrick Browne, Belinda S. Hall, Christopher Bot, Vithurshaa Gobalakrishnapillai, Peter F. Searle, Richard J. Knox, Shane R. Wilkinson
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 13) pp:3986-3991
Publication Date(Web):1 July 2011
DOI:10.1016/j.bmcl.2011.05.009
A series of nitrobenzyl phosphoramide mustards and their analogs was designed and synthesized to explore their structure–activity relationships as substrates of nitroreductases from Escherichia coli and trypanosomes and as potential antiproliferative and antiparasitic agents. The position of the nitro group on the phenyl ring was important with the 4-nitrobenzyl phosphoramide mustard (1) offering the best combination of enzyme activity and antiproliferative effect against both mammalian and trypanosomatid cells. A preference was observed for halogen substitutions ortho to benzyl phosphoramide mustard but distinct differences were found in their SAR of substituted 4-nitrobenzyl phosphoramide mustards in E. coli nitroreductase-expressing cells and in trypanosomatids expressing endogenous nitroreductases.
Co-reporter:Yongying Jiang, Robert S. DiPaola, Longqin Hu
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 9) pp:2587-2590
Publication Date(Web):1 May 2009
DOI:10.1016/j.bmcl.2009.03.009
In an effort to develop proteolytically activated prodrugs of phosphoramide mustard by prostate-specific antigen (PSA), a series of tetrapeptide (Cbz-Ser-Ser-Phe-Tyr)-conjugated 4-aminocyclophosphamide (4-NH2-CPA) isomers were synthesized and evaluated as substrates of PSA. The cleavage of the conjugates by PSA were found to be stereoselective as only the two isomers with 4R-configuration were efficiently cleaved by PSA. The cis-(2R,4R)-isomer was the best substrate of PSA with a half-life of 12 min. LC/MS analysis of the incubation solution of this isomer with PSA suggests that 4-NH2-CPA is released upon proteolysis and quickly degrades to cytotoxic phosphoramide mustard. These results clarified the stereochemical requirements of PSA on the peptide conjugates of 4-NH2-CPA and demonstrated the potential of these conjugates as potential PSA-activated prodrugs targeting prostate cancer.
Co-reporter:Yiyu Ge, Xinghua Wu, Dazhi Zhang, Longqin Hu
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 3) pp:941-944
Publication Date(Web):1 February 2009
DOI:10.1016/j.bmcl.2008.11.097
A novel linker system based on 3-aminoxypropionate was designed and evaluated for drug release using proteolysis as an activation trigger followed by intramolecular cyclization. The hydroxylamine moiety present in the linker system enabled faster release of the parent drug from the linker–drug conjugate at lower pH as compared to an aliphatic amine moiety. Introduction of two methyl groups strategically at the α position to the carboxylate in the linker further improved the rate of cyclization by nearly 2-fold. The 3-aminoxypropionate linker was successfully applied to a model prodrug for protease activation using α-chymotrypsin as the activating enzyme; the activation of the model prodrug bearing the 3-aminoxypropionate linker was 136 times faster than the corresponding model prodrug bearing an amine linker.
Co-reporter:Xinghua Wu, Yu Chen, Longqin Hu
Tetrahedron Letters 2009 50(40) pp: 5585-5588
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.07.080
Co-reporter:Yongying Jiang, Longqin Hu
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 14) pp:4059-4063
Publication Date(Web):15 July 2008
DOI:10.1016/j.bmcl.2008.05.099
N-(2,2-Dimethyl-2-(2-nitrophenyl)acetyl)-4-aminocyclophosphamide isomers (DMNA-NH-CPA, 4) were synthesized stereospecifically from Boc-l-Hse(OBn)-OH and the degradation of the corresponding reduced amine 5a was investigated by UV/vis spectroscopy and LC/MS. The rate of cyclization of 5a was found to increase with decreasing pH, with half-lives ranging from 3.2 to 54 min at pH 4–7.4, suggesting that the cyclization is catalyzed by the hydronium ions. LC/MS analysis of the degradation products of 5a indicates that 4-aminocyclophosphamide is rapidly released from 4 upon reductive activation under acidic conditions and further decomposes into the cytotoxic phosphoramide mustard. These results validated 4-aminocyclophosphamide as a prodrug form of phosphoramide mustard and suggest that compound 4 can potentially be used as a prodrug of phosphoramide mustard for bioreductive activation.N-(2,2-Dimethyl-2-(2-nitrophenyl)acetyl)-4-aminocyclophosphamide isomers were synthesized as potential bioreductively activated prodrugs of phosphoramide mustard and their mechanism of reductive activation was investigated.
Co-reporter:Yan-hui Yang, Herve Aloysius, Daigo Inoyama, Yu Chen, Long-qin Hu
Acta Pharmaceutica Sinica B (October 2011) Volume 1(Issue 3) pp:143-159
Publication Date(Web):October 2011
DOI:10.1016/j.apsb.2011.08.001
Co-reporter:Dhulfiqar Ali Abed, Melanie Goldstein, Haifa Albanyan, Huijuan Jin, Longqin Hu
Acta Pharmaceutica Sinica B (July 2015) Volume 5(Issue 4) pp:285-299
Publication Date(Web):July 2015
DOI:10.1016/j.apsb.2015.05.008

![4-Oxazolidinecarboxylicacid,3-[(2S)-2-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-1-oxopropyl]-2,2-dimethyl-,(4S)-](http://img.cochemist.com/ccimg/252600/252554-78-2.png)
![4-Oxazolidinecarboxylicacid,3-[(2S)-2-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-1-oxopropyl]-2,2-dimethyl-,(4S)-](http://img.cochemist.com/ccimg/252600/252554-78-2_b.png)


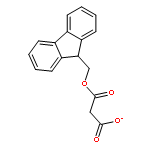
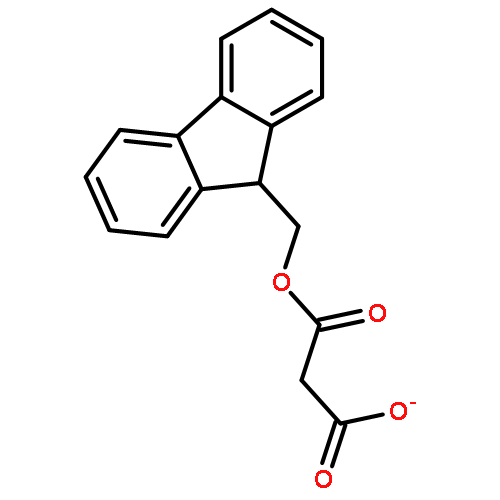
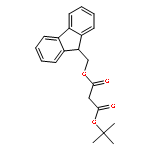
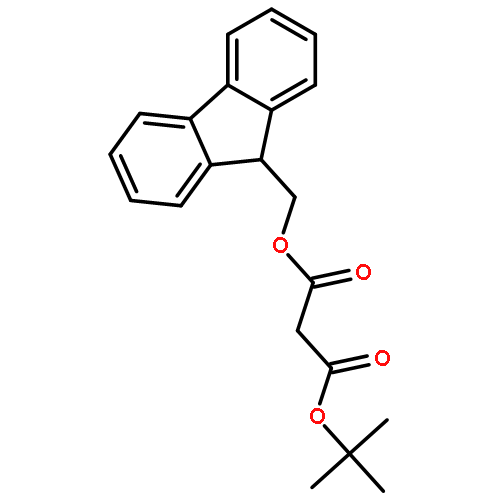




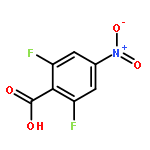
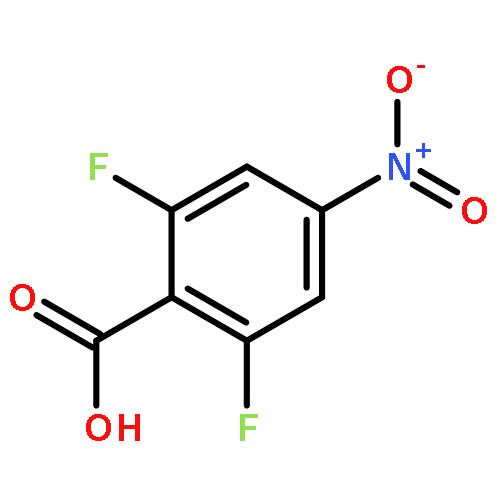
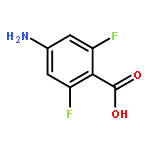
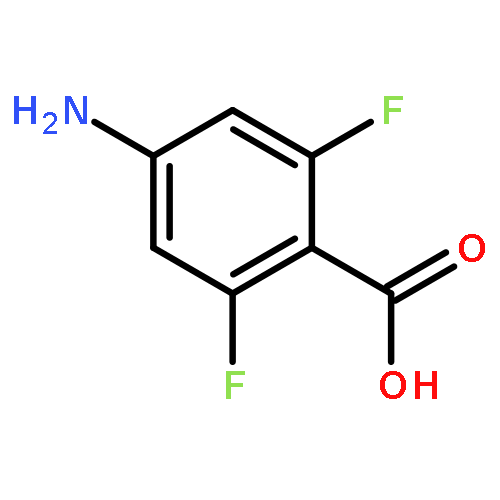
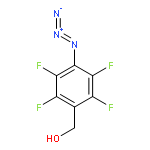
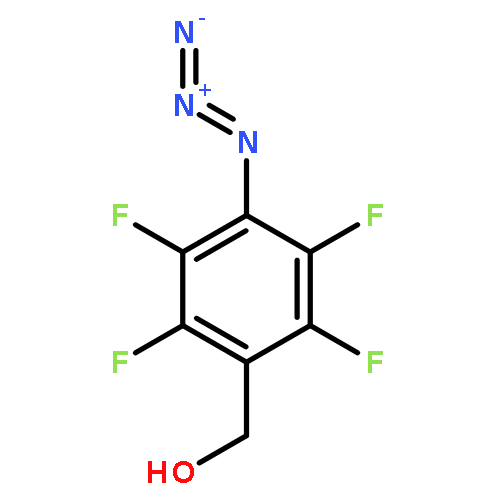
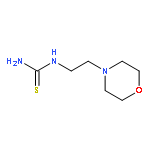
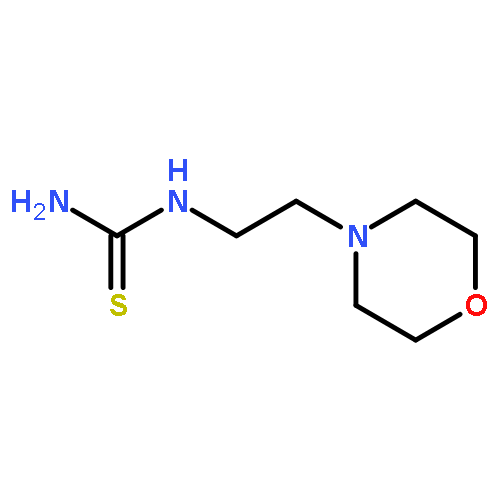
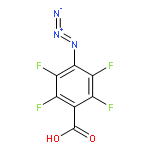
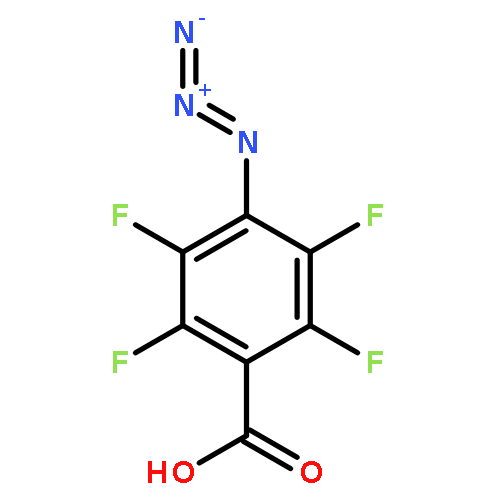
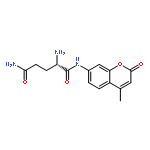
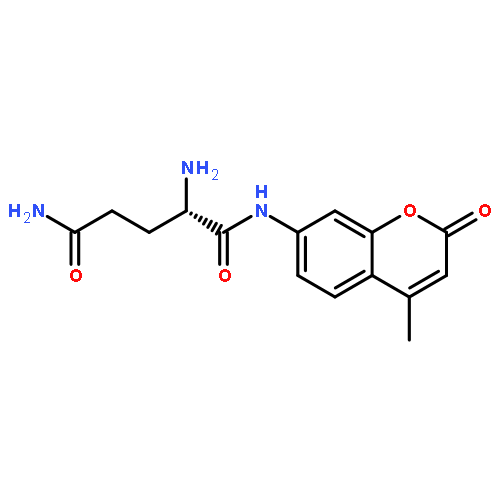
![Carbamic acid,[4-amino-1-[[(4-methyl-2-oxo-2H-1-benzopyran-7-yl)amino]carbonyl]-4-oxobutyl]-, 1,1-dimethylethyl ester, (S)-](http://img.cochemist.com/ccimg/105900/105888-43-5.png)
![Carbamic acid,[4-amino-1-[[(4-methyl-2-oxo-2H-1-benzopyran-7-yl)amino]carbonyl]-4-oxobutyl]-, 1,1-dimethylethyl ester, (S)-](http://img.cochemist.com/ccimg/105900/105888-43-5_b.png)
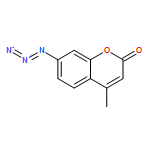
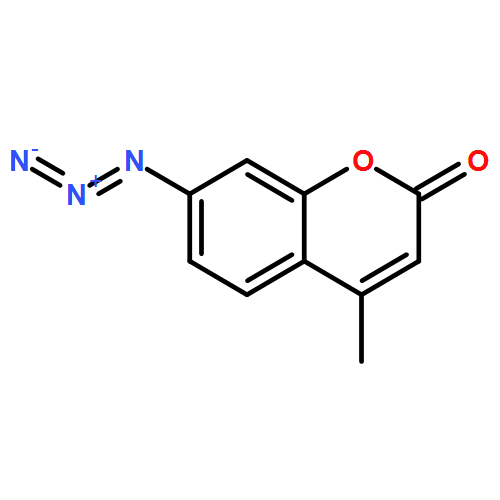
![(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate](http://img.cochemist.com/ccimg/67600/67526-95-8.png)
![(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate](http://img.cochemist.com/ccimg/67600/67526-95-8_b.png)
![Thiourea,N-[(tetrahydro-2-furanyl)methyl]-](http://img.cochemist.com/ccimg/66900/66892-25-9.png)
![Thiourea,N-[(tetrahydro-2-furanyl)methyl]-](http://img.cochemist.com/ccimg/66900/66892-25-9_b.png)