Co-reporter:P-C Chu, M-C Yang, S K Kulp, S B Salunke, L E Himmel, C-S Fang, A M Jadhav, Y-S Shan, C-T Lee, M-D Lai, L A Shirley, T Bekaii-Saab and C-S Chen
Oncogene 2016 35(30) pp:3897-3908
Publication Date(Web):November 30, 2015
DOI:10.1038/onc.2015.458
Integrin-linked kinase (ILK) is a mediator of aggressive phenotype in pancreatic cancer. On the basis of our finding that knockdown of either KRAS or ILK has a reciprocal effect on the other’s expression, we hypothesized the presence of an ILK-KRAS regulatory loop that enables pancreatic cancer cells to regulate KRAS expression. This study aimed to elucidate the mechanism by which this regulatory circuitry is regulated and to investigate the translational potential of targeting ILK to suppress oncogenic KRAS signaling in pancreatic cancer. Interplay between KRAS and ILK and the roles of E2F1, c-Myc and heterogeneous nuclear ribonucleoprotein as intermediary effectors in this feedback loop was interrogated by genetic manipulations through small interfering RNA/short hairpin RNA knockdown and ectopic expression, western blotting, PCR, promoter-luciferase reporter assays, chromatin immunoprecipitation and pull-down analyses. In vivo efficacy of ILK inhibition was evaluated in two murine xenograft models. Our data show that KRAS regulated the expression of ILK through E2F1-mediated transcriptional activation, which, in turn, controlled KRAS gene expression via hnRNPA1-mediated destabilization of the G-quadruplex on the KRAS promoter. Moreover, ILK inhibition blocked KRAS-driven epithelial–mesenchymal transition and growth factor-stimulated KRAS expression. The knockdown or pharmacological inhibition of ILK suppressed pancreatic tumor growth, in part, by suppressing KRAS signaling. These studies suggest that this KRAS-E2F1-ILK-hnRNPA1 regulatory loop enables pancreatic cancer cells to promote oncogenic KRAS signaling and to interact with the tumor microenvironment to promote aggressive phenotypes. This regulatory loop provides a mechanistic rationale for targeting ILK to suppress oncogenic KRAS signaling, which might foster new therapeutic strategies for pancreatic cancer.
Co-reporter:Ribai Yan; Hsiao-Ching Chuang; Naval Kapuriya; Chih-Chien Chou; Po-Ting Lai; Hsin-Wen Chang; Chia-Ning Yang; Samuel K. Kulp
Journal of Medicinal Chemistry 2015 Volume 58(Issue 5) pp:2290-2298
Publication Date(Web):February 17, 2015
DOI:10.1021/jm501751b
Previously, we reported that Akt inactivation by γ-tocopherol (2) in PTEN-negative prostate cancer cells resulted from its unique ability to facilitate membrane co-localization of Akt and PHLPP1 (PH domain leucine-rich repeat protein phosphatase isoform 1), a Ser473-specific Akt phosphatase, through pleckstrin homology (PH) domain binding. This finding provided a basis for exploiting 2 to develop a novel class of PHLPP1-targeted Akt inhibitors. Here, we used 3 (γ-VE5), a side chain-truncated 2 derivative, as a scaffold for lead optimization. The proof-of-concept of this structural optimization was obtained by 20, which exhibited higher antitumor efficacy than 3 in PTEN-negative cancer cells through PHLPP1-facilitated Akt inactivation. Like 3, 20 preferentially recognized the PH domains of Akt and PHLPP1, as its binding affinities for other PH domains, including those of ILK and PDK1, were an order-of-magnitude lower. Moreover, 20 was orally active in suppressing xenograft tumor growth in nude mice, which underlines the translational potential of this new class of Akt inhibitor in PTEN-deficient cancers.
Co-reporter:Santosh B. Salunke, Abul K. Azad, Naval P. Kapuriya, Joan-Miquel Balada-Llasat, Preeti Pancholi, Larry S. Schlesinger, Ching-Shih Chen
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 9) pp:1935-1943
Publication Date(Web):1 May 2015
DOI:10.1016/j.bmc.2015.03.041
The identification of compounds with anti-mycobacterial activity within classes of molecules that have been developed for other purposes is a fruitful approach for the development of anti-tuberculosis (TB) agents. In this study we used the scaffold of celecoxib which exhibits several activities against different pathogens, for the design and focused synthesis of a library of 64 compounds. For the primary screen, we used a bioluminescence-based method by constructing a luciferase-expressing reporter M.tb strain which contains the entire bacterial Lux operon cloned in a mycobacterial integrative expression vector. Through the screening of this library, we identified 6 hit compounds with high in vitro anti-mycobacterial activity (IC50 ∼0.18–0.48 μM). In particular, compounds 41, 51 and 53 were capable of inhibiting M.tb as effectively as the anti-TB drug isoniazid (INH) at 5 μM over a 72-h period, as analyzed by both bioluminescence- and colony forming unit (CFU)-based assays. All hit compounds also showed anti-M.tb activities against several multi-drug-resistant (MDR) strains. Most of the hit compounds showed no cytotoxicity for human macrophages at concentrations as high as 40 μM, setting the stage for further optimization and development of these anti-TB hit compounds both ex vivo and in vivo.
Co-reporter:Chih-Chien Chou;Santosh B. Salunke;Samuel K. Kulp
Journal of Cellular Biochemistry 2014 Volume 115( Issue 4) pp:611-624
Publication Date(Web):
DOI:10.1002/jcb.24704
ABSTRACT
Although the Human Genome Project has raised much hope for the identification of druggable genetic targets for cancer and other diseases, this genetic target-based approach has not improved productivity in drug discovery over the traditional approach. Analyses of known human target proteins of currently marketed drugs reveal that these drugs target only a limited number of proteins as compared to the whole proteome. In contrast to genome-based targets, mechanistic targets are derived from empirical research, at cellular or molecular levels, in disease models and/or in patients, thereby enabling the exploration of a greater number of druggable targets beyond the genome and epigenome. The paradigm shift has made a tremendous headway in developing new therapeutic agents targeting different clinically relevant mechanisms/pathways in cancer cells. In this Prospects article, we provide an overview of potential drug targets related to the following four emerging areas: (1) tumor metabolism (the Warburg effect), (2) dysregulated protein turnover (E3 ubiquitin ligases), (3) protein–protein interactions, and (4) unique DNA high-order structures and protein–DNA interactions. Nonetheless, considering the genetic and phenotypic heterogeneities that characterize cancer cells, the development of drug resistance in cancer cells by adapting signaling circuitry to take advantage of redundant pathways or feedback/crosstalk systems is possible. This “phenotypic adaptation” underlies the rationale of using therapeutic combinations of these targeted agents with cytotoxic drugs. J. Cell. Biochem. 115: 611–624, 2014. © 2013 Wiley Periodicals, Inc.
Co-reporter:Po-Hsien Huang;Hsiao-Ching Chuang;Huiling Wang;Hsiao-Ching Yang;Su-Lin Lee;Chih-Chien Chou;Naval Kapuriya;Dasheng Wang;Samuel K. Kulp;Hao-Chieh Chiu
Science Signaling 2013 Volume 6(Issue 267) pp:ra19
Publication Date(Web):19 Mar 2013
DOI:10.1126/scisignal.2003816
Vitamin E suppresses the proliferation of prostate cancer cells by inhibiting the growth-promoting kinase Akt.
Co-reporter:Dasheng Wang ; Po-Chen Chu ; Chia-Ning Yang ; Ribai Yan ; Yu-Chung Chuang ; Samuel K. Kulp
Journal of Medicinal Chemistry 2012 Volume 55(Issue 8) pp:3827-3836
Publication Date(Web):April 2, 2012
DOI:10.1021/jm300015m
On the basis of our finding that the antitumor effect of 5-{4-[(1-methylcyclohexyl)methoxy]benzyl}thiazolidine-2,4-dione, a thiazolidinedione peroxisome proliferator-activated receptor (PPAR)γ agonist, was, in part, attributable to its ability to block glucose uptake independently of PPARγ, we used its PPARγ-inactive analogue to develop a novel class of glucose transporter (GLUT) inhibitors. This lead optimization led to compound 30 {5-(4-hydroxy-3-trifluoromethylbenzylidene)-3-[4,4,4-trifluoro-2-methyl-2-(2,2,2-trifluoroethyl)butyl]thiazolidine-2,4-dione} as the optimal agent, which exhibited high antitumor potency through the suppression of glucose uptake (IC50, 2.5 μM), while not cytotoxic to prostate and mammary epithelial cells. This glucose uptake inhibition was associated with the inhibition of GLUT1 (IC50, 2 μM). Moreover, the mechanism of antitumor action of compound 30 was validated by its effect on a series of energy restriction-associated cellular responses. Homology modeling analysis suggests that the inhibitory effect of compound 30 on glucose entry was attributable to its ability to bind to the GLUT1 channel at a site distinct from that of glucose.
Co-reporter:Hao-Chieh Chiu, Su-Lin Lee, Naval Kapuriya, Dasheng Wang, Yi-Ru Chen, Sung-Liang Yu, Samuel K. Kulp, Lee-Jene Teng, Ching-Shih Chen
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 15) pp:4653-4660
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmc.2012.06.018
Methicillin-resistant Staphylococcus aureus (MRSA) poses a serious threat to public health because of its resistance to multiple antibiotics most commonly used to treat infection. In this study, we report the unique ability of the cyclooxygenase-2 (COX-2) inhibitor celecoxib to kill Staphylococcus aureus and MRSA with modest potency. We hypothesize that the anti-Staphylococcus activity of celecoxib could be pharmacologically exploited to develop novel anti-MRSA agents with a distinct mechanism. Examination of an in-house, celecoxib-based focused compound library in conjunction with structural modifications led to the identification of compound 46 as the lead agent with high antibacterial potency against a panel of Staphylococcus pathogens and different strains of MRSA. Moreover, this killing effect is bacteria-specific, as human cancer cells are resistant to 46. In addition, a single intraperitoneal administration of compound 46 at 30 mg/kg improved the survival of MRSA-infected C57BL/6 mice. In light of its high potency in eradicating MRSA in vitro and its in vivo activity, compound 46 and its analogues warrant continued preclinical development as a potential therapeutic intervention against MRSA.
Co-reporter:Su-Lin Lee ; En-Chi Hsu ; Chih-Chien Chou ; Hsiao-Ching Chuang ; Li-Yuan Bai ; Samuel K. Kulp
Journal of Medicinal Chemistry 2011 Volume 54(Issue 18) pp:6364-6374
Publication Date(Web):August 8, 2011
DOI:10.1021/jm2007744
Integrin-linked kinase (ILK) represents a relevant target for cancer therapy in light of its role in promoting oncogenesis and tumor progression. Through the screening of an in-house focused compound library, we identified N-methyl-3-(1-(4-(piperazin-1-yl)phenyl)-5-(4′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)-1H-pyrazol-3-yl)propanamide (22) as a novel ILK inhibitor (IC50, 0.6 μM), which exhibited high in vitro potency against a panel of prostate and breast cancer cell lines (IC50, 1–2.5 μM), while normal epithelial cells were unaffected. Compound 22 facilitated the dephosphorylation of Akt at Ser-473 and other ILK targets, including glycogen synthase kinase-3β and myosin light chain. Moreover, 22 suppressed the expression of the transcription/translation factor YB-1 and its targets HER2 and EGFR in PC-3 cells, which could be rescued by the stable expression of constitutively active ILK. Evidence indicates that 22 induced autophagy and apoptosis, both of which were integral to its antiproliferative activity. Together, this broad spectrum of mechanisms underlies the therapeutic potential of 22 in cancer treatment, which is manifested by its in vivo efficacy as a single oral agent in suppressing PC-3 xenograft tumor growth.
Co-reporter:Jih-Hwa Guh ; Wei-Ling Chang ; Jian Yang ; Su-Lin Lee ; Shuo Wei ; Dasheng Wang ; Samuel K. Kulp
Journal of Medicinal Chemistry 2010 Volume 53(Issue 6) pp:2552-2561
Publication Date(Web):February 19, 2010
DOI:10.1021/jm901773d
In light of the unique ability of thiazolidinediones to mediate peroxisome proliferator-activated receptor (PPAR)γ-independent activation of adenosine monophosphate-activated protein kinase (AMPK) and suppression of interleukin (IL)-6 production, we conducted a screening of an in-house, thiazolidinedione-based focused compound library to identify novel agents with these dual pharmacological activities. Cell-based assays pertinent to the activation status of AMPK and mammalian homologue of target of rapamycin (i.e., phosphorylation of AMPK and p70 ribosomal protein S6 kinase, respectively) and IL-6/IL-6 receptor signaling (i.e., IL-6 production and signal transducer and activator of transcription 3 phosphorylation, respectively) in lipopolysaccharide (LPS)-stimulated THP-1 human macrophages were used to screen this compound library, which led to the identification of compound 53 (N-{4-[3-(1-methyl-cyclohexylmethyl)-2,4-dioxo-thiazolidin-5-ylidene-methyl]-phenyl}-4-nitro-3-trifluoro-methyl-benzenesulfonamide) as the lead agent. Evidence indicates that this drug-induced suppression of LPS-stimulated IL-6 production was attributable to AMPK activation. Furthermore, compound 53-mediated AMPK activation was demonstrated in C-26 colon adenocarcinoma cells, indicating that it is not a cell line-specific event.
Co-reporter:Dasheng Wang ; Hsiao-Ching Chuang ; Shu-Chuan Weng ; Po-Hsien Huang ; Hao-Yu Hsieh ; Samuel K. Kulp
Journal of Medicinal Chemistry 2009 Volume 52(Issue 18) pp:5642-5648
Publication Date(Web):August 26, 2009
DOI:10.1021/jm9002457
This study is aimed at the pharmacological exploitation of α-tocopheryl succinate (1) to develop potent antiadhesion agents. Considering the structural cooperativity between the phytyl chain and the carboxylic terminus in determining the antiadhesion activity, our structural optimization led to compound 5 ([2-(4,8-dimethyl-non-1-enyl)-2,5,7,8-tetramethyl-chroman-6-yloxy]-acetic acid), which exhibited an-order-of-magnitude higher potency than 1 in blocking the adhesion of 4T1 metastatic breast cancer cells to extracellular matrix proteins (IC50, 0.6 μM versus 10 μM). Evidence indicates that the ability of compound 5 to block cell adhesion and migration was attributable to its effect on disrupting focal adhesion and actin cytoskeletal integrity by facilitating the degradation of focal adhesion kinase. Interactions between tumor cells and the ECM in the tumor microenvironment have been increasingly recognized as critical modulators of the metastatic potential of tumor cells. Consequently, the ability of compound 5 to block such interactions provides a unique pharmacological tool to shed light onto mechanisms that govern cell adhesion and tumor metastasis.
Co-reporter:Hao-Chieh Chiu;Shilpa Soni;Samuel K Kulp;Heather Curry
Journal of Biomedical Science 2009 Volume 16( Issue 1) pp:
Publication Date(Web):2009 December
DOI:10.1186/1423-0127-16-110
Autophagy has been shown recently to play an important role in the intracellular survival of several pathogenic bacteria. In this study, we investigated the effect of a novel small-molecule autophagy-inducing agent, AR-12, on the survival of Francisella tularensis, the causative bacterium of tularemia in humans and a potential bioterrorism agent, in macrophages.Our results show that AR-12 induces autophagy in THP-1 macrophages, as indicated by increased autophagosome formation, and potently inhibits the intracellular survival of F. tularensis (type A strain, Schu S4) and F. novicida in macrophages in association with increased bacterial co-localization with autophagosomes. The effect of AR-12 on intracellular F. novicida was fully reversed in the presence of the autophagy inhibitor, 3-methyl adenine or the lysosome inhibitor, chloroquine. Intracellular F. novicida were not susceptible to the inhibitory activity of AR-12 added at 12 h post-infection in THP-1 macrophages, and this lack of susceptibility was independent of the intracellular location of bacteria.Together, AR-12 represents a proof-of-principle that intracellular F. tularensis can be eradicated by small-molecule agents that target innate immunity.
Co-reporter:Jian Yang ; Shuo Wei ; Da-Sheng Wang ; Yu-Chieh Wang ; Samuel K. Kulp
Journal of Medicinal Chemistry 2008 Volume 51(Issue 7) pp:2100-2107
Publication Date(Web):March 13, 2008
DOI:10.1021/jm701212m
On the basis of our finding that the peroxisome proliferator-activated receptor γ (PPARγ) agonist ciglitazone at high doses was able to mediate PPARγ-independent transcriptional repression of androgen receptor (AR) in a tumor cell-specific manner, we used Δ2CG, a PPARγ-inactive analogue of ciglitazone, to conduct lead optimization to develop a novel class of AR-ablative agents. Structure–activity analysis indicates a high degree of flexibility in realigning Δ2CG’s structural moieties without compromising potency in AR repression, as evidenced by the higher AR-ablative activity of the permuted isomer 9 [(Z)-5-(4-hydroxybenzylidene)-3-(1-methylcyclohexylmethyl)thiazolidine-2,4-dione]. Further modificiations of 9 gave rise to 12 [(Z)-5-(4-hydroxy-3-trifluoromethylbenzylidene)-3-(1-methylcyclohexylmethyl)thiazolidine-2,4-dione], which completely inhibited AR expression in LNCaP cells at low micromolar concentrations. This AR down-regulation led to growth inhibition in LNCaP cells through apoptosis induction. Moreover, the role of AR repression in the antiproliferative effect of compound 12 was validated by the differential inhibition of cell viability between androgen-responsive and androgen-nonresponsive cells.
Co-reporter:Da-Sheng Wang, Ching-Shih Chen
Bioorganic & Medicinal Chemistry 2001 Volume 9(Issue 12) pp:3165-3172
Publication Date(Web):December 2001
DOI:10.1016/S0968-0896(01)00232-2
We have synthesized a series of 3-deoxy-3-heteromethyl derivatives of l-α-phosphatidyl-d-myo-inositol as part of our effort to develop specific, reversible inhibitors of phosphoinositide (PI) 3-kinase. Among various derivatives examined, phosphatidyl-d-3-deoxy-3-aminomethyl-myo-inositol displays the highest potency in inhibiting PI 3-kinase both in vitro and in cells. It effectively suppressed antigen-stimulated degranulation in mast cells (IC50, 17 μM), suggesting a potential application of this PI 3-kinase inhibitor as a mast cell-stabilizing agent.Graphic
Co-reporter:En-Chi Hsu, Samuel K. Kulp, Han-Li Huang, Huang-Ju Tu, ... Ching-Shih Chen
Neoplasia (June 2015) Volume 17(Issue 6) pp:497-508
Publication Date(Web):1 June 2015
DOI:10.1016/j.neo.2015.06.001
Interleukin-6 (IL-6) and Notch signaling are important regulators of breast cancer stem cells (CSCs), which drive the malignant phenotype through self-renewal, differentiation, and development of therapeutic resistance. We investigated the role of integrin-linked kinase (ILK) in regulating IL-6–driven Notch1 activation and the ability to target breast CSCs through ILK inhibition. Ectopic expression/short hairpin RNA-mediated knockdown of ILK, pharmacological inhibition of ILK with the small molecule T315, Western blot analysis, immunofluorescence, and luciferase reporter assays were used to evaluate the regulation of IL-6–driven Notch1 activation by ILK in IL-6–producing triple-negative breast cancer cell lines (MDA-MB-231, SUM-159) and in MCF-7 and MCF-7IL-6 cells. The effects of ILK on γ-secretase complex assembly and cellular localization were determined by immunofluorescence, Western blots of membrane fractions, and immunoprecipitation. In vivo effects of T315-induced ILK inhibition on CSCs in SUM-159 xenograft models were assessed by mammosphere assays, flow cytometry, and tumorigenicity assays. Results show that the genetic knockdown or pharmacological inhibition of ILK suppressed Notch1 activation and the abundance of the γ-secretase components presenilin-1, nicastrin, and presenilin enhancer 2 at the posttranscriptional level via inhibition of caveolin-1-dependent membrane assembly of the γ-secretase complex. Accordingly, knockdown of ILK inhibited breast CSC-like properties in vitro and the breast CSC subpopulation in vivo in xenograft tumor models. Based on these findings, we propose a novel function of ILK in regulating γ-secretase–mediated Notch1 activation, which suggests the targeting of ILK as a therapeutic approach to suppress IL-6–induced breast CSCs.
Co-reporter:Ya-Ting Yang, Curt Balch, Samuel K. Kulp, Michael R. Mand, ... Ching-Shih Chen
Neoplasia (June 2009) Volume 11(Issue 6) pp:552-563- IN7-IN9
Publication Date(Web):1 June 2009
DOI:10.1593/neo.09204
Histone deacetylase inhibitors (HDACIs) are a class of antineoplastic agents previously demonstrating preclinical chemosensitizing activity against drug-resistant cancer cells and mouse xenografts. However, whereas clinical studies have shown efficacy against human hematologic malignancies, solid tumor trials have proved disappointing. We previously developed a novel HDACI, “OSU-HDAC42,” and herein examine its activity against ovarian cancer cell lines and xenografts. OSU-HDAC42, (i) unlike most HDACIs, elicited a more than five-fold increase in G2-phase cells, at 2.5 µM, with G2 arrest followed by apoptosis; (ii) at 1.0 µM, completely repressed messenger RNA expression of the cell cycle progression gene cdc2; (iii) at low doses (0.25–1.0 µM for 24 hours), induced tumor cell epithelial differentiation, as evidenced by morphology changes and a more than five-fold up-regulation of epithelium-specific cytokeratins; (iv) potently abrogated the growth of numerous ovarian cancer cells, with IC50 values of 0.5 to 1.0 µM, whereas also remaining eight-fold less toxic (IC50 of 8.6 µM) to normal ovarian surface epithelial cells; and (v) chemosensitizated platinum-resistant mouse xenografts to cisplatin. Compared with the clinically approved HDACI suberoylanilide hydroxamic acid (vorinostat), 1.0 µM OSU-HDAC42 was more biochemically potent (i.e., enzyme-inhibitory), as suggested by greater gene up-regulation and acetylation of both histone and nonhistone proteins. In p53-dysfunctional cells, however, OSU-HDAC42 was two- to eight-fold less inductive of p53-regulated genes, whereas also having a two-fold higher IC50 than p53-functional cells, demonstrating some interaction with p53 tumor-suppressive cascades. These findings establish OSU-HDAC42 as a promising therapeutic agent for drug-resistant ovarian cancer and justify its further investigation.
Co-reporter:Sally E. Henderson, Li-Yun Ding, Xiaokui Mo, Tanios Bekaii-Saab, ... Po-Hsien Huang
Neoplasia (December 2016) Volume 18(Issue 12) pp:765-774
Publication Date(Web):1 December 2016
DOI:10.1016/j.neo.2016.10.003
PURPOSE: Pancreatic ductal adenocarcinoma (PDAC) is the third leading cause of cancer death in the United States. This study was aimed at evaluating the efficacy of AR-42 (formerly OSU-HDAC42), a novel histone deacetylase (HDAC) inhibitor currently in clinical trials, in suppressing tumor growth and/or cancer-induced muscle wasting in murine models of PDAC. EXPERIMENTAL DESIGN: The in vitro antiproliferative activity of AR-42 was evaluated in six human pancreatic cancer cell lines (AsPC-1, COLO-357, PANC-1, MiaPaCa-2, BxPC-3, SW1990). AsPC-1 subcutaneous xenograft and transgenic KPfl/flC (LSL-KrasG12D;Trp53flox/flox;Pdx-1-Cre) mouse models of pancreatic cancer were used to evaluate the in vivo efficacy of AR-42 in suppressing tumor growth and/or muscle wasting. RESULTS: Growth suppression in AR-42–treated cells was observed in all six human pancreatic cancer cell lines with dose-dependent modulation of proliferation and apoptotic markers, which was associated with the hallmark features of HDAC inhibition, including p21 upregulation and histone H3 hyperacetylation. Oral administration of AR-42 at 50 mg/kg every other day resulted in suppression of tumor burden in the AsPC-1 xenograft and KPfl/flC models by 78% and 55%, respectively, at the end of treatment. Tumor suppression was associated with HDAC inhibition, increased apoptosis, and inhibition of proliferation. Additionally, AR-42 as a single agent preserved muscle size and increased grip strength in KPfl/flC mice. Finally, the combination of AR-42 and gemcitabine in transgenic mice demonstrated a significant increase in survival than either agent alone. CONCLUSIONS: These results suggest that AR-42 represents a therapeutically promising strategy for the treatment of pancreatic cancer.
Co-reporter:Wei-Ling Chang, Chia-Chun Yu, Ching-Shih Chen, Jih-Hwa Guh
Biochemical Pharmacology (1 March 2015) Volume 94(Issue 1) pp:12-21
Publication Date(Web):1 March 2015
DOI:10.1016/j.bcp.2015.01.005





![3-(1H-Indol-3-yl)-4-[2-(4-methylpiperazin-1-yl)quinazolin-4-yl]pyrrole-2,5-dione](http://img.cochemist.com/ccimg/425700/425637-18-9.png)
![3-(1H-Indol-3-yl)-4-[2-(4-methylpiperazin-1-yl)quinazolin-4-yl]pyrrole-2,5-dione](http://img.cochemist.com/ccimg/425700/425637-18-9_b.png)
![Ethanone, 1-[3-(bromomethyl)phenyl]-2,2,2-trifluoro-](http://img.cochemist.com/ccimg/370200/370104-05-5.png)
![Ethanone, 1-[3-(bromomethyl)phenyl]-2,2,2-trifluoro-](http://img.cochemist.com/ccimg/370200/370104-05-5_b.png)
































![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6_b.png)
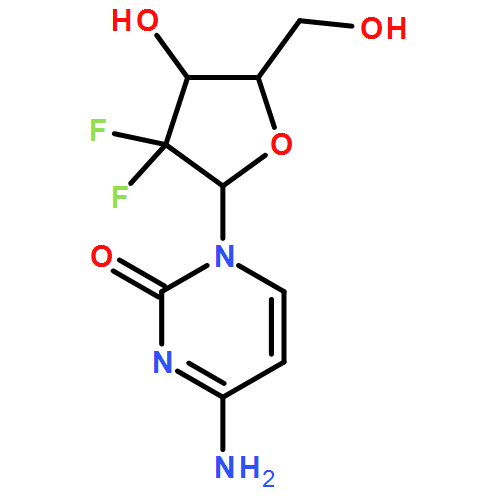
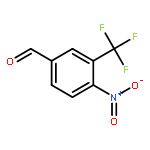
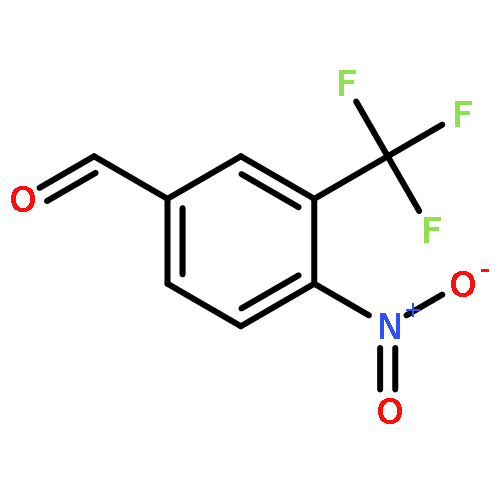


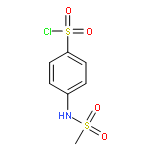
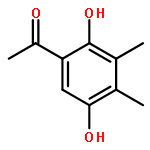
![ETHANONE, 1-[4-(BROMOMETHYL)PHENYL]-2,2,2-TRIFLUORO-](http://img.cochemist.com/ccimg/78200/78126-14-4.png)
![ETHANONE, 1-[4-(BROMOMETHYL)PHENYL]-2,2,2-TRIFLUORO-](http://img.cochemist.com/ccimg/78200/78126-14-4_b.png)
![[4-(4-AMINO-6,7-DIMETHOXY-QUINAZOLIN-2- YL)PIPERAZIN-1-YL]-(2,5-DIOXABI CYCLO[4.4.0]DECA-6,8,10-TRIEN-4-YL)METHANONE](http://img.cochemist.com/ccimg/74200/74191-85-8.png)
![[4-(4-AMINO-6,7-DIMETHOXY-QUINAZOLIN-2- YL)PIPERAZIN-1-YL]-(2,5-DIOXABI CYCLO[4.4.0]DECA-6,8,10-TRIEN-4-YL)METHANONE](http://img.cochemist.com/ccimg/74200/74191-85-8_b.png)
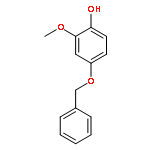
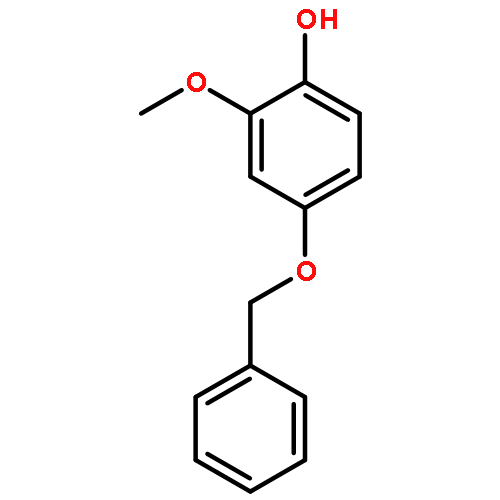
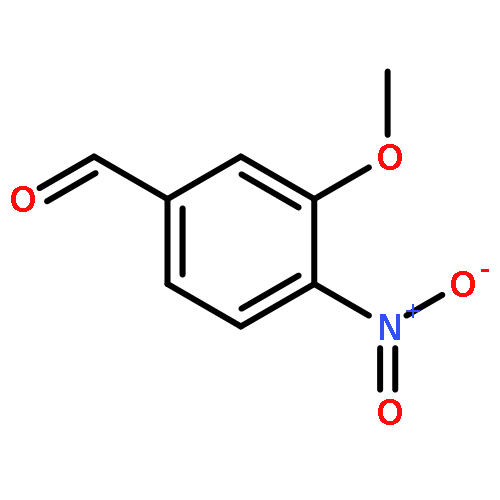
![Benzenesulfonyl chloride, 4-[(trifluoromethyl)thio]-](http://img.cochemist.com/ccimg/67300/67216-75-5.png)
![Benzenesulfonyl chloride, 4-[(trifluoromethyl)thio]-](http://img.cochemist.com/ccimg/67300/67216-75-5_b.png)
![2,4-Thiazolidinedione,5-[(4-nitrophenyl)methylene]-](http://img.cochemist.com/ccimg/34400/34301-40-1.png)
![2,4-Thiazolidinedione,5-[(4-nitrophenyl)methylene]-](http://img.cochemist.com/ccimg/34400/34301-40-1_b.png)


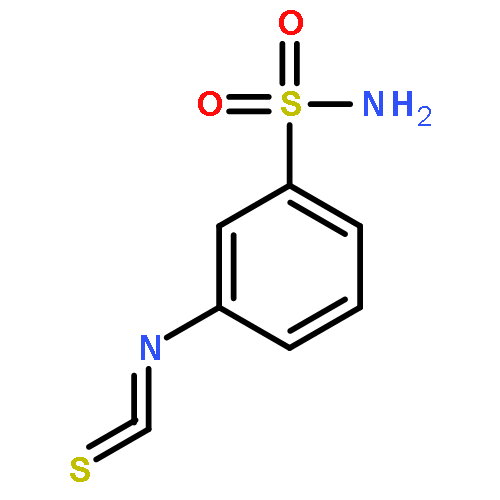
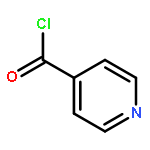
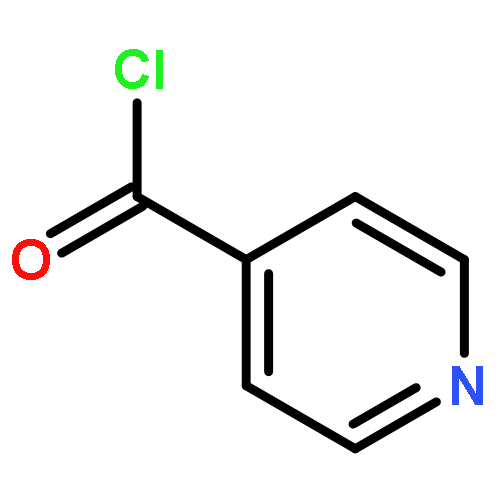
![Indolo[2',3':3,4]pyrido[2,1-b]quinazolin-5(7H)-one,8,13,13b,14-tetrahydro-14-methyl- (6CI,9CI)](http://img.cochemist.com/ccimg/6000/5956-87-6.png)
![Indolo[2',3':3,4]pyrido[2,1-b]quinazolin-5(7H)-one,8,13,13b,14-tetrahydro-14-methyl- (6CI,9CI)](http://img.cochemist.com/ccimg/6000/5956-87-6_b.png)