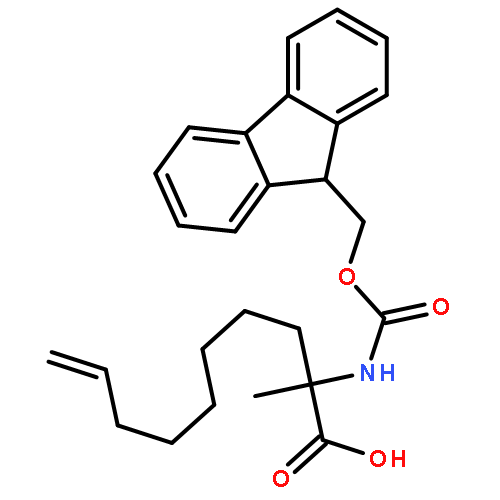
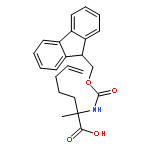
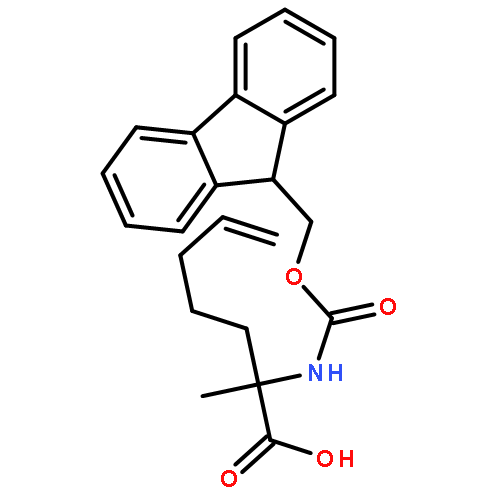
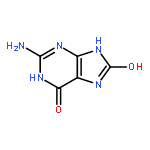
Co-reporter:Khian Hong Pua, Dylan T. Stiles, Mathew E. Sowa, Gregory L. Verdine
Cell Reports 2017 Volume 18, Issue 2(Volume 18, Issue 2) pp:
Publication Date(Web):10 January 2017
DOI:10.1016/j.celrep.2016.12.030
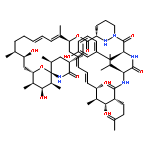
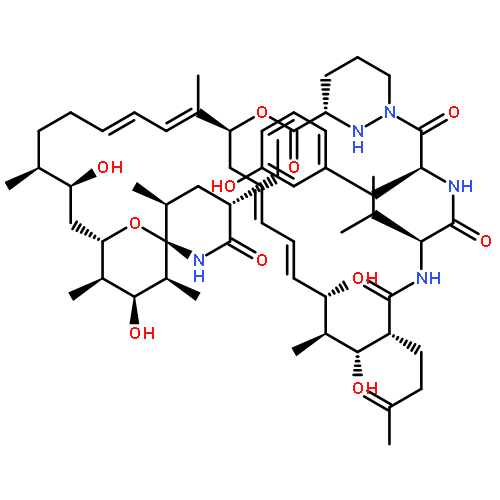
•SFA engages IMPDH2 in a PPIA-dependent manner•PPIA-SFA interaction with IMPDH is highly isoform specific•PPIA-SFA complex binds the CBS domains of IMPDH2•CBS domains of IMPDH2 are implicated in cellular proliferationNatural products have demonstrated utility in the clinic and can also act as probes to understand complex cellular pathways. Sanglifehrin A (SFA) is a mixed polyketide and non-ribosomal peptide synthase natural product with sub-nano-molar affinity for its receptor cyclophilin A (PPIA). It has been shown to behave in vitro as an immune suppressant. Here, we identify inosine-5′-monophosphate dehydrogenase 2 (IMPDH2) as an intracellular target of the PPIA-SFA binary complex. The formation of this ternary complex does not inhibit the enzymatic activity of IMPDH2. Rather, ternary complex formation modulates cell growth through interaction with the cystathionine-β-synthase (CBS) domain of IMPDH2. We further demonstrate that the SFA complex is highly isoform selective for IMPDH2 (versus IMPDH1). This work reveals a role for the CBS domains of IMPDH2 in cellular proliferation, suggesting a more complex role than previously suspected for IMPDH2 in T cell activation and proliferation.Download high-res image (264KB)Download full-size image
Co-reporter:Qian Chu, Raymond E. Moellering, Gerard J. Hilinski, Young-Woo Kim, Tom N. Grossmann, Johannes T.-H. Yeh and Gregory L. Verdine
MedChemComm 2015 vol. 6(Issue 1) pp:111-119
Publication Date(Web):14 Oct 2014
DOI:10.1039/C4MD00131A
Hydrocarbon-stapled α-helical peptides are a new class of targeting molecules capable of penetrating cells and engaging intracellular targets formerly considered intractable. This technology has been applied to the development of cell-permeable ligands targeting key intracellular protein–protein interactions. However, the properties governing cell penetration of hydrocarbon-stapled peptides have not yet been rigorously investigated. Herein we report our studies to systematically probe cellular uptake of stapled peptides. We developed a high-throughput epifluorescence microscopy assay to quantitatively measure stapled peptide intracellular accumulation and demonstrated that this assay yielded highly reproducible results. Using this assay, we analyzed more than 200 peptides with various sequences, staple positions and types, and found that cell penetration ability is strongly related to staple type and formal charge, whereas other physicochemical parameters do not appear to have a significant effect. We next investigated the mechanism(s) involved in stapled peptide internalization and have demonstrated that stapled peptides penetrate cells through a clathrin- and caveolin-independent endocytosis pathway that involves, in part, sulfated cell surface proteoglycans, but that also seems to exploit a novel, uncharacterized pathway. Taken together, staple type and charge are the key physical properties in determining the cell penetration ability of stapled peptides, and anionic cell surface proteoglycans might serve as receptors to mediate stapled peptide internalization. These findings improve our understanding of stapled peptides as chemical probes and potential targeted therapeutics, and provide useful guidelines for the design of next-generation stapled peptides with enhanced cell permeability.
Co-reporter:Gerard J. Hilinski ; Young-Woo Kim ; Jooyeon Hong ; Peter S. Kutchukian ; Charisse M. Crenshaw ; Shaunna S. Berkovitch ; Andrew Chang ; Sihyun Ham
Journal of the American Chemical Society 2014 Volume 136(Issue 35) pp:12314-12322
Publication Date(Web):August 8, 2014
DOI:10.1021/ja505141j
Conformationally stabilized α-helical peptides are capable of inhibiting disease-relevant intracellular or extracellular protein–protein interactions in vivo. We have previously reported that the employment of ring-closing metathesis to introduce a single all-hydrocarbon staple along one face of an α-helical peptide greatly increases α-helical content, binding affinity to a target protein, cell penetration through active transport, and resistance to proteolytic degradation. In an effort to improve upon this technology for stabilizing a peptide in a bioactive α-helical conformation, we report the discovery of an efficient and selective bis ring-closing metathesis reaction leading to peptides bearing multiple contiguous staples connected by a central spiro ring junction. Circular dichroism spectroscopy, NMR, and computational analyses have been used to investigate the conformation of these “stitched” peptides, which are shown to exhibit remarkable thermal stabilities. Likewise, trypsin proteolysis assays confirm the achievement of a structural rigidity unmatched by peptides bearing a single staple. Furthermore, fluorescence-activated cell sorting (FACS) and confocal microscopy assays demonstrate that stitched peptides display superior cell penetrating ability compared to their stapled counterparts, suggesting that this technology may be useful not only in the context of enhancing the drug-like properties of α-helical peptides but also in producing potent agents for the intracellular delivery of proteins and oligonucleotides.
Co-reporter:So Youn Shim;Young-Woo Kim
Chemical Biology & Drug Design 2013 Volume 82( Issue 6) pp:635-642
Publication Date(Web):
DOI:10.1111/cbdd.12231
We have previously shown that the incorporation of an 8-atom all-hydrocarbon ‘staple’ at positions i and i + 3 of a synthetic peptide results in substantial stabilization of the α-helical conformation. As part of our ongoing effort to explore the scope and utility of all-hydrocarbon stapling systems, we have investigated and report herein the properties of a new i, i + 3 stapling system that employs a 6-carbon cross-link.
Co-reporter:Brian R. Bowman;Tom N. Grossmann;Johannes T.-H. Yeh;Raymond E. Moellering;Qian Chu
PNAS 2012 Volume 109 (Issue 44 ) pp:17942-17947
Publication Date(Web):2012-10-30
DOI:10.1073/pnas.1208396109
Aberrant activation of signaling by the Wnt pathway is strongly implicated in the onset and progression of numerous types
of cancer. Owing to the persistent dependence of these tumors on Wnt signaling for growth and survival, inhibition of this
pathway is considered an attractive mechanism-based therapeutic approach. Oncogenic activation of Wnt signaling can ensue
from a variety of distinct aberrations in the signaling pathway, but most share the common feature of causing increased cellular
levels of β-catenin by interfering with its constitutive degradation. β-Catenin serves as a central hub in Wnt signaling by
engaging in crucial protein–protein interactions with both negative and positive effectors of the pathway. Direct interference
with these protein–protein interactions is a biologically compelling approach toward suppression of β-catenin hyperactivity,
but such interactions have proven intransigent with respect to small-molecule targeting. Hence β-catenin remains an elusive
target for translational cancer therapy. Here we report the discovery of a hydrocarbon-stapled peptide that directly targets
β-catenin and interferes with its ability to serve as a transcriptional coactivator for T-cell factor (TCF) proteins, the
downstream transcriptional regulators of the Wnt pathway.
Co-reporter:Yan Qi;Marie C. Spong;Kwangho Nam;Anirban Banerjee;Rou-Jia Sung;Martin Karplus;Michael Zhang
PNAS 2012 Volume 109 (Issue 4 ) pp:
Publication Date(Web):2012-01-24
DOI:10.1073/pnas.1111237108
Base excision repair of genotoxic nucleobase lesions in the genome is critically dependent upon the ability of DNA glycosylases
to locate rare sites of damage embedded in a vast excess of undamaged DNA, using only thermal energy to fuel the search process.
Considerable interest surrounds the question of how DNA glycosylases translocate efficiently along DNA while maintaining their
vigilance for target damaged sites. Here, we report the observation of strandwise translocation of 8-oxoguanine DNA glycosylase,
MutM, along undamaged DNA. In these complexes, the protein is observed to translocate by one nucleotide on one strand while
remaining untranslocated on the complementary strand. We further report that alterations of single base-pairs or a single
amino acid substitution (R112A) can induce strandwise translocation. Molecular dynamics simulations confirm that MutM can
translocate along DNA in a strandwise fashion. These observations reveal a previously unobserved mode of movement for a DNA-binding
protein along the surface of DNA.
Co-reporter:Young-Woo Kim, Peter S. Kutchukian and Gregory L. Verdine
Organic Letters 2010 Volume 12(Issue 13) pp:3046-3049
Publication Date(Web):June 7, 2010
DOI:10.1021/ol1010449
The introduction of all-hydrocarbon i,i+3 staples into α-helical peptide scaffolds via ring-closing olefin metathesis (RCM) between two α-methyl,α-pentenylglycine residues incorporated at i and i+3 positions, which lie on the same face of the helix, has been investigated. The reactions were found to be highly dependent upon the side-chain stereochemistry of the amino acids undergoing RCM. The i,i+3 stapling system established here provides a potentially useful alternative to the well-established i,i+4 stapling system now in widespread use.
Co-reporter:Yan Qi,
Marie C. Spong,
Kwangho Nam,
Anirban Banerjee,
Sao Jiralerspong,
Martin Karplus
&
Gregory L. Verdine
Nature 2009 462(7274) pp:762
Publication Date(Web):2009-12-10
DOI:10.1038/nature08561
In living systems, the repair of genotoxic damage requires that the lesion first be detected in an excess of undamaged DNA. A base-excision DNA repair enzyme, MutM, is now captured and structurally elucidated at the stage of initial encounter with a damaged nucleobase within a DNA duplex. By combining structural biology and computational modelling, the pathway by which this encounter causes the damaged nucleobase to be extruded from the DNA duplex is defined.
Co-reporter:Raymond E. Moellering,
Melanie Cornejo,
Tina N. Davis,
Cristina Del Bianco,
Jon C. Aster,
Stephen C. Blacklow,
Andrew L. Kung,
D. Gary Gilliland,
Gregory L. Verdine
&
James E. Bradner
Nature 2009 462(7270) pp:182
Publication Date(Web):2009-11-12
DOI:10.1038/nature08543
It is notoriously difficult to target transcription factors with aberrant activity in cancer. Inappropriate activation of the NOTCH complex of transcription factors is directly implicated in the pathogenesis of several disease states, including T-cell acute lymphoblastic leukaemia. The design of synthetic, cell-permeable, stabilized -helical peptides that disrupt protein–protein interactions in NOTCH is now described.
Co-reporter:Seongmin Lee
PNAS 2009 Volume 106 (Issue 44 ) pp:18497-18502
Publication Date(Web):2009-11-03
DOI:10.1073/pnas.0902908106
Adenine DNA glycosylase catalyzes the glycolytic removal of adenine from the promutagenic A·oxoG base pair in DNA. The general
features of DNA recognition by an adenine DNA glycosylase, Bacillus stearothermophilus MutY, have previously been revealed via the X-ray structure of a catalytically inactive mutant protein bound to an A:oxoG-containing
DNA duplex. Although the structure revealed the substrate adenine to be, as expected, extruded from the DNA helix and inserted
into an extrahelical active site pocket on the enzyme, the substrate adenine engaged in no direct contacts with active site
residues. This feature was paradoxical, because other glycosylases have been observed to engage their substrates primarily
through direct contacts. The lack of direct contacts in the case of MutY suggested that either MutY uses a distinctive logic
for substrate recognition or that the X-ray structure had captured a noncatalytically competent state in lesion recognition.
To gain further insight into this issue, we crystallized wild-type MutY bound to DNA containing a catalytically inactive analog
of 2′-deoxyadenosine in which a single 2′-H atom was replaced by fluorine. The structure of this fluorinated lesion-recognition
complex (FLRC) reveals the substrate adenine buried more deeply into the active site pocket than in the prior structure and
now engaged in multiple direct hydrogen bonding and hydrophobic interactions. This structure appears to capture the catalytically
competent state of adenine DNA glycosylases, and it suggests a catalytic mechanism for this class of enzymes, one in which
general acid-catalyzed protonation of the nucleobase promotes glycosidic bond cleavage.
Co-reporter:Anirban Banerjee;Webster L. Santos
Science 2006 Vol 311(5764) pp:1153-1157
Publication Date(Web):24 Feb 2006
DOI:10.1126/science.1120288
Abstract
DNA glycosylases must interrogate millions of base pairs of undamaged DNA in order to locate and then excise one damaged nucleobase. The nature of this search process remains poorly understood. Here we report the use of disulfide cross-linking (DXL) technology to obtain structures of a bacterial DNA glycosylase, MutM, interrogating undamaged DNA. These structures, solved to 2.0 angstrom resolution, reveal the nature of the search process: The protein inserts a probe residue into the helical stack and severely buckles the target base pair, which remains intrahelical. MutM therefore actively interrogates the intact DNA helix while searching for damage.
Co-reporter:Gregory L. Verdine, Gerard J. Hilinski
Drug Discovery Today: Technologies (Spring 2012) Volume 9(Issue 1) pp:e41-e47
Publication Date(Web):1 March 2012
DOI:10.1016/j.ddtec.2012.01.004
A majority of proteins responsible for the establishment and maintenance of human disease states are unable to be targeted therapeutically by molecules belonging to either of the two established classes of drugs, namely small molecules and protein therapeutics. Recent efforts toward drugging these ‘undruggable’ proteins have led to greatly increased focus on cell-penetrating mini-proteins as a new class of agents for targeting intractable intracellular proteins. These molecules are designed to combine the advantages intrinsic to each conventional therapeutic modality while overcoming their individual limitations. One class of cell-penetrating mini-proteins, all-hydrocarbon stapled peptides, has recently demonstrated the ability to potently and specifically target previously intractable proteins such as transcription factors, vaulting this class to the forefront of a new wave of next-generation drugs.
Co-reporter:Brian R. Bowman, Seongmin Lee, Shuyu Wang, Gregory L. Verdine
Structure (6 August 2008) Volume 16(Issue 8) pp:1166-1174
Publication Date(Web):6 August 2008
DOI:10.1016/j.str.2008.04.012
The constant attack on DNA by endogenous and exogenous agents gives rise to nucleobase modifications that cause mutations, which can lead to cancer. Visualizing the effects of these lesions on the structure of duplex DNA is key to understanding their biologic consequences. The most definitive method of obtaining such structures, X-ray crystallography, is troublesome to employ owing to the difficulty of obtaining diffraction-quality crystals of DNA. Here, we present a crystallization system that uses a protein, the DNA glycosylase AlkA, as a scaffold to mediate the crystallization of lesion-containing duplex DNA. We demonstrate the use of this system to facilitate the rapid structure determination of DNA containing the lesion 8-oxoguanine in several different sequence contexts, and also deoxyinosine and 1,N6-ethenoadenine, each stabilized as the corresponding 2′-flouro analog. The structures of 8-oxoguanine provide a correct atomic-level view of this important endogenous lesion in DNA.
Co-reporter:Danaya Pakotiprapha, Yoshihiko Inuzuka, Brian R. Bowman, Geri F. Moolenaar, ... Gregory L. Verdine
Molecular Cell (18 January 2008) Volume 29(Issue 1) pp:122-133
Publication Date(Web):18 January 2008
DOI:10.1016/j.molcel.2007.10.026
The nucleotide excision repair pathway corrects many structurally unrelated DNA lesions. Damage recognition in bacteria is performed by UvrA, a member of the ABC ATPase superfamily whose functional form is a dimer with four nucleotide-binding domains (NBDs), two per protomer. In the 3.2 Å structure of UvrA from Bacillus stearothermophilus, we observe that the nucleotide-binding sites are formed in an intramolecular fashion and are not at the dimer interface as is typically found in other ABC ATPases. UvrA also harbors two unique domains; we show that one of these is required for interaction with UvrB, its partner in lesion recognition. In addition, UvrA contains three zinc modules, the number and ligand sphere of which differ from previously published models. Structural analysis, biochemical experiments, surface electrostatics, and sequence conservation form the basis for models of ATP-modulated dimerization, UvrA-UvrB interaction, and DNA binding during the search for lesions.