Co-reporter:Mohammad B. Khaled, Roukaya K. El Mokadem, and Jimmie D. Weaver III
Journal of the American Chemical Society September 20, 2017 Volume 139(Issue 37) pp:13092-13092
Publication Date(Web):August 24, 2017
DOI:10.1021/jacs.7b06847
The photocatalytic C–F functionalization of highly fluorinated arenes is a powerful method for accessing functionalized multifluorinated arenes. The decisive step in the determining regioselectivity in fluorine functionalization is fluoride fragmentation from the radical anion of the multifluorinated arene. To date, the availability of regioisomers has been dictated by the innate electronics of the fluorinated arene, limiting the synthetic utility of the chemistry. This study investigates the remarkable ability of a strategically located hydrogen bond to transcend the normal regioselectivity of the C–F functionalization event. A significant rate acceleration is additionally observed for hydrodefluorination of fluorines that can undergo intramolecular hydrogen bonds that form 5–8-membered cycles with moderately acidic N–H’s. The hydrogen bond is expected to facilitate the fragmentation not only by bending the C–F bond of the radical anion out of planarity but also by increasing the exothermicity of the fluoride extrusion step through protonation of the naked fluoride. Finally, the synthetic utility of the method is demonstrated in an expedited synthesis of the trifluorinated α-phenyl acetic acid derivative required for the commercial synthesis of Januvia, an antidiabetic drug. This represents the first synthesis of a commercially important multifluorinated arene via a defluorination strategy and is significantly shorter than the current strategy.
Co-reporter:Jon I. Day and Jimmie D. Weaver
The Journal of Organic Chemistry July 7, 2017 Volume 82(Issue 13) pp:6801-6801
Publication Date(Web):June 9, 2017
DOI:10.1021/acs.joc.7b00962
Functionalized per- and polyfluoroarenes are important building blocks, with many industrially and medicinally important molecules containing them. Nucleophilic aromatic substitution can be employed as a quick and straightforward way to synthesize these building blocks. While many methods to derivatize fluoroarenes exist that use heteroatom centered nucleophiles, there are fewer methods that use carbon centered nucleophiles, and of those many are poorly defined. This work presents the SNAr reaction of nucleophiles generated from nitroalkanes with a variety of fluorinated arenes. Given that the products are versatile, accessing polyfluorinated arene building blocks in substantial scale is important. This method is highly regioselective, and produces good to moderate yields on a large scale, sans chromatography, and thus fulfills this need. In addition, the regioselectivity of the addition was probed using both DFT calculations and experimentally via halogen exchange.
Co-reporter:Kip A. Teegardin;Jimmie D. Weaver
Chemical Communications 2017 vol. 53(Issue 35) pp:4771-4774
Publication Date(Web):2017/04/27
DOI:10.1039/C7CC01606A
Herein, conditions are provided for the formation and use of the oxazolone enolate for the nucleophilic substitution of highly fluorinated (hetero)arenes, which after unmasking yield highly fluorinated non-natural amino acids and derivatives. In addition, the properties and chemical behavior of this new class of amino acids are explored. The utility is demonstrated in the one pot synthesis of medicinally relevant 2-aminohydantoins.
Co-reporter:Sameera Senaweera;Jimmie D. Weaver
Chemical Communications 2017 vol. 53(Issue 54) pp:7545-7548
Publication Date(Web):2017/07/04
DOI:10.1039/C7CC03996D
Selective catalytic SNAr reaction of polyfluoroaryl C–F bonds with chloride is shown. Stoichiometric TMSCl makes the reaction exergonic and allows catalysis, which involves ground state elevation of chloride, aromatic donor–acceptor interactions, and stabilization of the Meisenheimer complex. Traditional cross-coupling of the products is now possible and demonstrates the utility.
Co-reporter:Amandeep Arora and Jimmie D. Weaver
Accounts of Chemical Research 2016 Volume 49(Issue 10) pp:2273
Publication Date(Web):September 28, 2016
DOI:10.1021/acs.accounts.6b00259
Photocatalysis offers several mechanistically unique pathways that are not rivaled by mainstream catalysis. Primarily, the ability to convert photochemical energy into single electron oxidation and reduction events provides a new dimension for chemists to consider when choosing how to activate a molecule or approach a complex synthesis. Since most organic molecules do not absorb light in the visible region, they are impervious to direct visible light photochemistry, which provides an opportunity for photocatalysis in which a visible light absorbing compound can serve as a mediator.In this Account, we discuss the consequences of catalyst mediated, photoinduced electron transfer to several classes of reducible arenes. While the bulk of the work discussed within this Account utilizes iridium-based photocatalysts, in principle the chemistry is not limited to this class of photocatalyst, and the principles should be more general. Instead, this Account focuses largely on the consequences of single electron transfer to poly- and perfluorinated arenes and 2-halo azoles. Electron transfer converts these stable molecules into reactive intermediates whose behavior often depends entirely on the identity of the halogen that undergoes substitution. The result is both diverse chemistry and an alternative way of thinking about the chemical reactivity of these motifs. Specifically, we discuss our efforts and those of others to develop strategies for the generation of radicals or radical anions from perfluoroarenes and azoles and the behavior of these intermediates as implied by reactions in which they participate. The divergent pathway is illustrated by 2-bromoazoles, which yield azolyl radicals and can be utilized for addition to π-bonds, while use of the 2-chloroazole substrate leads to an entirely different reaction profile. Under the appropriate reaction conditions, the reactive and transient intermediates are useful coupling partners and often provide unrivaled access to new chemical space. The odd electron species can form challenging bonds with minimal prefunctionalization of the coupling partner. For instance, some of the intermediates can be utilized for C–H functionalizations to selectively make crowded amines or to synthesize biarenes substituted at every ortho position. While photocatalysis is not the only manner of accomplishing electron transfer, the catalytic generation of the reactive species in which the concentration of the transient odd electron species is kept low, provides a synthetic handle that can be used to improve reaction outcomes. This is elegantly demonstrated in a number of examples in which redox sensitive groups located on substrates survive the reaction. In addition, the underlying basic concepts associated with radical anion fragmentation are reviewed and provide the backdrop for discussion throughout the Account.
Co-reporter:Sameera Senaweera;Jimmie D. Weaver
Journal of the American Chemical Society 2016 Volume 138(Issue 8) pp:2520-2523
Publication Date(Web):February 18, 2016
DOI:10.1021/jacs.5b13450
Multifluorinated biaryls are challenging to synthesize and yet are an important class of molecules. Because of the difficulty associated with selective fluorination, this class of molecules represent a formidable synthetic challenge. An alternative approach to selective fluorination of biaryls is to couple an arene that already possesses C–F bonds in the desired location. This strategy has been regularly utilized and relies heavily on traditional cross-coupling strategies that employ organometallics and halides (or pseudohalides) in order to achieve the coupling. Herein we report conditions for the photocatalytic coupling via direct functionalization of the C–F bond of a perfluoroarene and C–H bond of the other arene to provide an expedient route to multifluorinated biaryls. The mild conditions and good functional group tolerance enable a broad scope, including access to the anti-Minisci product of basic heterocycles. Finally, we demonstrate the value of the C–F functionalization approach by utilizing the high fluorine content to systematically build complex biaryls containing between two and five Caryl–F bonds via the synergistic use of photocatalysis and SNAr chemistry.
Co-reporter:Amandeep Arora and Jimmie D. Weaver
Organic Letters 2016 Volume 18(Issue 16) pp:3996-3999
Publication Date(Web):August 5, 2016
DOI:10.1021/acs.orglett.6b01718
The 2-azolyl radical, generated from 2-bromoazoles via photocatalysis, is a powerful intermediate for the intermolecular arylation of unmodified (hetero)arenes. The reaction is characterized by mild conditions, operational simplicity, tolerance toward functional and sterically demanding groups, broad scope, and anti-Minisci selectivity. A working mechanism is provided, and a low-solubility amine is essential for successful coupling. The utility of the reaction is demonstrated via late-stage functionalization of methyl estrone and application toward other bromoarenes.
Co-reporter:Kip Teegardin, Jon I. Day, John Chan, and Jimmie Weaver
Organic Process Research & Development 2016 Volume 20(Issue 7) pp:1156-1163
Publication Date(Web):June 28, 2016
DOI:10.1021/acs.oprd.6b00101
Photocatalytic organic transformations utilizing ruthenium and iridium complexes have garnered significant attention due to the access they provide to new synthetic spaces through new reaction mechanisms. A survey of the photophysical data and the diversity of transformations that may be accomplished utilizing commercially available photocatalysts is contained herein.
Co-reporter:A. Singh, J. J. Kubik and J. D. Weaver
Chemical Science 2015 vol. 6(Issue 12) pp:7206-7212
Publication Date(Web):29 Sep 2015
DOI:10.1039/C5SC03013G
C–F functionalizations that provide C–C bonds are challenging synthetic transformations, due in part to the large C–F bond strength, short bond length, nonpolarizable nature, the production of fluoride, and the regioselectivity-in the case of multifluorinated substrates. However, commercially available highly fluorinated arenes possess great synthetic potential because they already possess the C–F bonds in the desired locations that would be difficult to selectively fluorinate. In order to take advantage of this potential, selective C–F functionalizations must be developed. Herein, we disclose conditions for the photocatalytic reductive alkylation of highly fluorinated arenes with ubiquitous and unactivated alkenes. The mild reaction conditions provide for a broad functional group scope, and the reaction is remarkably efficient using just 0.25 mol% catalyst. Finally, we demonstrate the utility of the strategy by converting highly fluorinated arenes to elaborate (hetero)arenes that contain 2–5 Caryl–F bonds via synergistic use of photocatalysis and SNAr chemistry.
Co-reporter:Amandeep Arora, Kip A. Teegardin, and Jimmie D. Weaver
Organic Letters 2015 Volume 17(Issue 15) pp:3722-3725
Publication Date(Web):July 14, 2015
DOI:10.1021/acs.orglett.5b01711
Access to Csp2–Csp3-coupled products is a challenging goal at the forefront of catalysis. The photocatalytic reductive coupling of aryl bromides with unactivated alkenes is introduced as a convenient method that circumvents any need for synthesis of sp3-hybridized coupling partners. The reaction takes place via photoinduced electron transfer from a tertiary amine to an aryl bromide that fragments to provide an aryl radical and subsequently reacts with an alkene to form a C–C bond. Conveniently, the amine also serves as the final reductant. The method is operationally simple, functional group tolerant, and takes place with selectivities that will allow it to be used in the context of complex molecule synthesis.
Co-reporter:Anuradha Singh, Kip Teegardin, Megan Kelly, Kariate S. Prasad, Sadagopan Krishnan, Jimmie D. Weaver
Journal of Organometallic Chemistry 2015 Volume 776() pp:51-59
Publication Date(Web):15 January 2015
DOI:10.1016/j.jorganchem.2014.10.037
•Synthesis of iridium photocatalysts directly from IrCl3 in 1 step and high yields.•Determination of redox properties.•Determination of triplet state energies.•Spectral characterization.Herein we describe an improved synthesis for homoleptic iridium(III) 2-phenylpyridine based photocatalysts that allows rapid access to these compounds in good to high yields which have recently become a vital component within the field of catalysis. In addition, we synthesized a number of heteroleptic iridium(III) 2-phenylpyridine photocatalysts and report their photophysical and electrochemical properties. The emission energies span the range of 473–560 nm and reduction potentials from −2.27 V to −1.23 V and oxidation potentials ranging from 1.81 V to 0.69 V. Additionally, we provide the calculated excited state properties and comment on the role of these properties in designing catalytic cycles.The emission energies of the cyclometalated iridium-complexes span the range of 473–560 nm and are shown pictographically by their corresponding color. This range corresponds to a 9 kcal/mol energy difference in available triplet state energy.
Co-reporter:Sameera M. Senaweera ; Anuradha Singh ;Jimmie D. Weaver
Journal of the American Chemical Society 2014 Volume 136(Issue 8) pp:3002-3005
Publication Date(Web):February 18, 2014
DOI:10.1021/ja500031m
Polyfluorinated aromatics are essential to materials science as well as the pharmaceutical and agrochemical industries and yet are often difficult to access. This Communication describes a photocatalytic hydrodefluorination approach which begins with easily accessible perfluoroarenes and selectively reduces the C–F bonds. The method allows facile access to a number of partially fluorinated arenes and takes place with unprecedented catalytic activity using a safe and inexpensive amine as the reductant.
Co-reporter:Kamaljeet Singh ; Shannon J. Staig ;Jimmie D. Weaver
Journal of the American Chemical Society 2014 Volume 136(Issue 14) pp:5275-5278
Publication Date(Web):March 28, 2014
DOI:10.1021/ja5019749
Catalytic access to thermodynamically less stable Z-alkenes has recently received considerable attention. These approaches have relied upon kinetic control of the reaction to arrive at the thermodynamically less stable geometrical isomer. Herein, we present an orthogonal approach which proceeds via photochemically catalyzed isomerization of the thermodynamic E-alkene to the less stable Z-isomer which occurs via a photochemical pumping mechanism. We consider two potential mechanisms. Importantly, the reaction conditions are mild, tolerant, and operationally simple and will be easily implemented.
Co-reporter:Sameera M. Senaweera and Jimmie D. Weaver
The Journal of Organic Chemistry 2014 Volume 79(Issue 21) pp:10466-10476
Publication Date(Web):October 1, 2014
DOI:10.1021/jo502075p
This work describes the facile and mono-selective per- and polyfluoroarylation of Meldrum’s acid to generate a versatile synthon for highly fluorinated α-phenyl acetic acid derivatives, which provide straightforward access to fluorinated building blocks. The reaction takes place quickly, and most products were isolated without the need for chromatography. Importantly, this method provides an alternative strategy to access α-arylated Meldrum’s acids, which avoids the need for aryl-Pb(IV) salts or diaryliodonium salts. Furthermore, we demonstrate the synthetic versatility and utility of the Meldrum’s acid products by subjecting our products to several derivatizations of the Meldrum’s acid products as well as photocatalytic hydrodefluorination.
Co-reporter:Anuradha Singh, Amandeep Arora, and Jimmie D. Weaver
Organic Letters 2013 Volume 15(Issue 20) pp:5390-5393
Publication Date(Web):October 7, 2013
DOI:10.1021/ol402751j
Herein, conditions for C–H functionalization of tertiary aliphatic amines and their subsequent coupling with a number of 2-chloroazole derivatives are reported. The reaction is facilitated by a catalytic amount of tris-fac-Ir(ppy)3, with blue light irradiation and takes place under mild and convenient conditions. Most couplings take place with excellent regioselectivity. The reaction is tolerant of a number of functional groups and allows for rapid access to α-azole carbinamines commonly found in post-translationally modified peptides.
Co-reporter:A. Singh, J. J. Kubik and J. D. Weaver
Chemical Science (2010-Present) 2015 - vol. 6(Issue 12) pp:NaN7212-7212
Publication Date(Web):2015/09/29
DOI:10.1039/C5SC03013G
C–F functionalizations that provide C–C bonds are challenging synthetic transformations, due in part to the large C–F bond strength, short bond length, nonpolarizable nature, the production of fluoride, and the regioselectivity-in the case of multifluorinated substrates. However, commercially available highly fluorinated arenes possess great synthetic potential because they already possess the C–F bonds in the desired locations that would be difficult to selectively fluorinate. In order to take advantage of this potential, selective C–F functionalizations must be developed. Herein, we disclose conditions for the photocatalytic reductive alkylation of highly fluorinated arenes with ubiquitous and unactivated alkenes. The mild reaction conditions provide for a broad functional group scope, and the reaction is remarkably efficient using just 0.25 mol% catalyst. Finally, we demonstrate the utility of the strategy by converting highly fluorinated arenes to elaborate (hetero)arenes that contain 2–5 Caryl–F bonds via synergistic use of photocatalysis and SNAr chemistry.
Co-reporter:Kip A. Teegardin and Jimmie D. Weaver
Chemical Communications 2017 - vol. 53(Issue 35) pp:NaN4774-4774
Publication Date(Web):2017/03/30
DOI:10.1039/C7CC01606A
Herein, conditions are provided for the formation and use of the oxazolone enolate for the nucleophilic substitution of highly fluorinated (hetero)arenes, which after unmasking yield highly fluorinated non-natural amino acids and derivatives. In addition, the properties and chemical behavior of this new class of amino acids are explored. The utility is demonstrated in the one pot synthesis of medicinally relevant 2-aminohydantoins.
Co-reporter:A. Singh, C. J. Fennell and J. D. Weaver
Chemical Science (2010-Present) 2016 - vol. 7(Issue 11) pp:NaN6802-6802
Publication Date(Web):2016/07/21
DOI:10.1039/C6SC02422J
Photocatalytic alkene synthesis can involve electron and energy transfer processes. The structure of the photocatalyst can be used to control the rate of the energy transfer, providing a mechanistic handle over the two processes. Jointly considering catalyst volume and emissive energy provides a highly sensitive strategy for predicting which mechanistic pathway will dominate. This model was developed en route to a photocatalytic Caryl–F alkenylation reaction of alkynes and highly-fluorinated arenes as partners. By judicious choice of photocatalyst, access to E- or Z-olefins was accomplished, even in the case of synthetically challenging trisubstituted alkenes. The generality and transferability of this model was tested by evaluating established photocatalytic reactions, resulting in shortened reaction times and access to complimentary Z-cinnamylamines in the photocatalytic [2 + 2] and C–H vinylation of amines, respectively. These results show that taking into account the size of the photocatalyst provides predictive ability and control in photochemical quenching events.
Co-reporter:Sameera Senaweera and Jimmie D. Weaver
Chemical Communications 2017 - vol. 53(Issue 54) pp:NaN7548-7548
Publication Date(Web):2017/06/21
DOI:10.1039/C7CC03996D
Selective catalytic SNAr reaction of polyfluoroaryl C–F bonds with chloride is shown. Stoichiometric TMSCl makes the reaction exergonic and allows catalysis, which involves ground state elevation of chloride, aromatic donor–acceptor interactions, and stabilization of the Meisenheimer complex. Traditional cross-coupling of the products is now possible and demonstrates the utility.










![2-Propen-1-ol, 3-[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/514800/514797-25-2.png)
![2-Propen-1-ol, 3-[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/514800/514797-25-2_b.png)



![Dibenzo[b,e][1,4]dioxin, 1,2-difluoro-](http://img.cochemist.com/ccimg/161600/161586-83-0.png)
![Dibenzo[b,e][1,4]dioxin, 1,2-difluoro-](http://img.cochemist.com/ccimg/161600/161586-83-0_b.png)
![Dibenzo[b,e][1,4]dioxin, 1,2,4-trifluoro-](http://img.cochemist.com/ccimg/161600/161586-65-8.png)
![Dibenzo[b,e][1,4]dioxin, 1,2,4-trifluoro-](http://img.cochemist.com/ccimg/161600/161586-65-8_b.png)
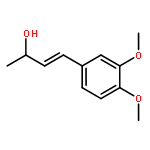
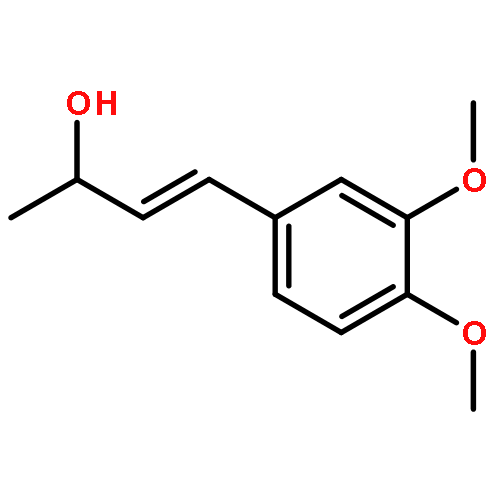
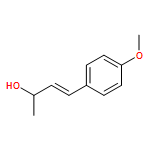
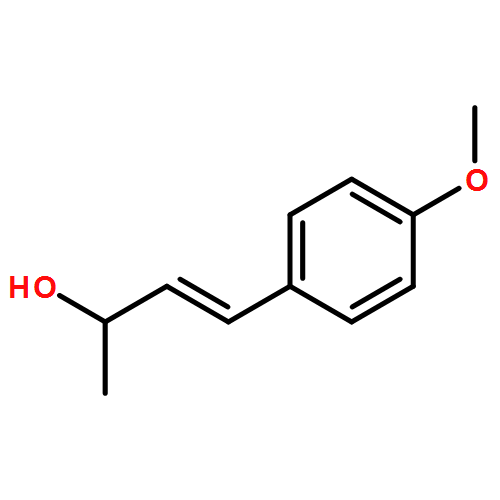



![Benzenamine, N-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/92600/92573-86-9.png)
![Benzenamine, N-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/92600/92573-86-9_b.png)
![MORPHOLINE, 4-[(2E)-3-PHENYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/85700/85620-82-2.png)
![MORPHOLINE, 4-[(2E)-3-PHENYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/85700/85620-82-2_b.png)





![PYRROLIDINE, 1-[(2E)-3-PHENYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/58400/58325-60-3.png)
![PYRROLIDINE, 1-[(2E)-3-PHENYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/58400/58325-60-3_b.png)


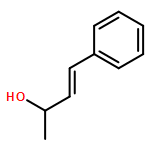
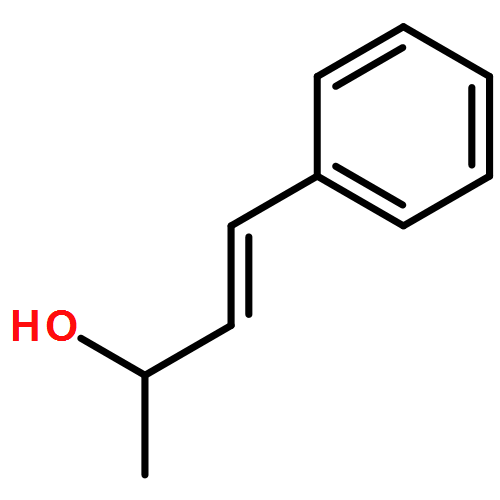
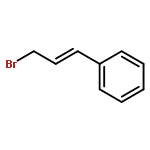
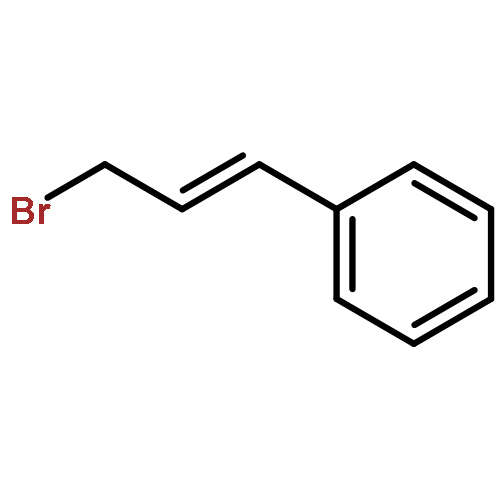




![Benzene, [(1E)-3-methyl-1-buten-1-yl]-](/data/chemimg/1744400/15325-61-8.png)
![Benzene, [(1E)-3-methyl-1-buten-1-yl]-](/data/chemimg/1744400/15325-61-8_b.png)
![Benzene, [(1Z)-3-methyl-1-buten-1-yl]-](/data/chemimg/1738300/15325-56-1.png)
![Benzene, [(1Z)-3-methyl-1-buten-1-yl]-](/data/chemimg/1738300/15325-56-1_b.png)


![Piperidine, 1-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/5900/5882-82-6.png)
![Piperidine, 1-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/5900/5882-82-6_b.png)
![1-{2-[(4-methyl-1,2,5-oxadiazol-3-yl)oxy]ethyl}-5-phenyl-1H-tetrazole](http://img.cochemist.com/ccimg/5900/5844-54-2.png)
![1-{2-[(4-methyl-1,2,5-oxadiazol-3-yl)oxy]ethyl}-5-phenyl-1H-tetrazole](http://img.cochemist.com/ccimg/5900/5844-54-2_b.png)



![Dimethyl (1r,2s,3r,4s)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate](http://img.cochemist.com/ccimg/2000/1984-43-6.png)
![Dimethyl (1r,2s,3r,4s)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate](http://img.cochemist.com/ccimg/2000/1984-43-6_b.png)
![1,5-Dioxaspiro[5.5]undecane-2,4-dione, 3-methyl-](http://img.cochemist.com/ccimg/1700/1658-28-2.png)
![1,5-Dioxaspiro[5.5]undecane-2,4-dione, 3-methyl-](http://img.cochemist.com/ccimg/1700/1658-28-2_b.png)
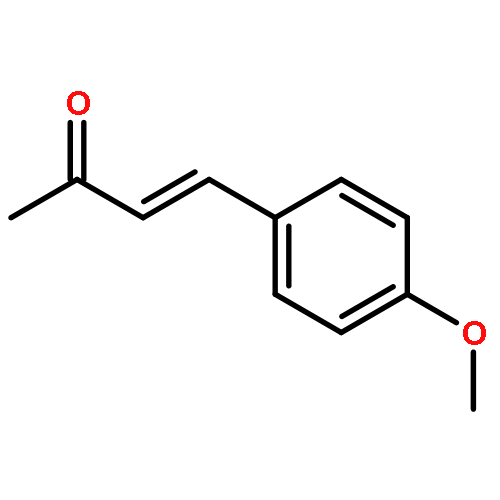
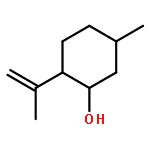
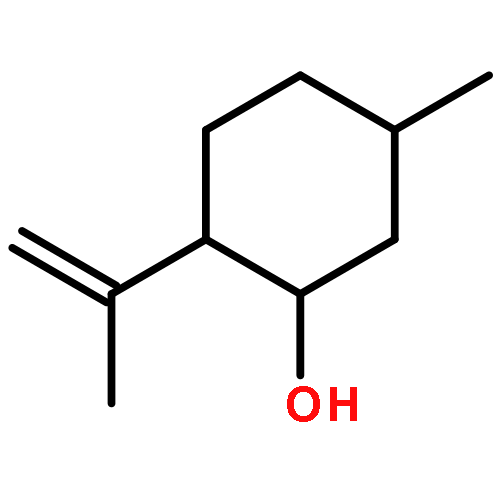
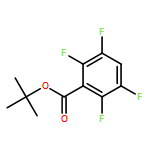


![Dibenzo[b,e][1,4]dioxin, 1,2,3,4-tetrafluoro-](http://img.cochemist.com/ccimg/119600/119546-24-6.png)
![Dibenzo[b,e][1,4]dioxin, 1,2,3,4-tetrafluoro-](http://img.cochemist.com/ccimg/119600/119546-24-6_b.png)
![N-[2,5-DIFLUORO-4-(TRIFLUOROMETHYL)PHENYL]ACETAMIDE](/data/chemimg/369500/114973-35-2.png)
![N-[2,5-DIFLUORO-4-(TRIFLUOROMETHYL)PHENYL]ACETAMIDE](/data/chemimg/369500/114973-35-2_b.png)
![1,5-Dioxaspiro[5.5]undecane-2,4-dione](http://img.cochemist.com/ccimg/1700/1658-27-1.png)
![1,5-Dioxaspiro[5.5]undecane-2,4-dione](http://img.cochemist.com/ccimg/1700/1658-27-1_b.png)