Co-reporter:Nandita Abhyankar, Jin Jung Kweon, Maylis Orio, Sylvain Bertaina, Minseong Lee, Eun Sang Choi, Riqiang Fu, and Naresh S. Dalal
The Journal of Physical Chemistry C March 23, 2017 Volume 121(Issue 11) pp:6314-6314
Publication Date(Web):February 20, 2017
DOI:10.1021/acs.jpcc.7b00596
Dimethylammonium zinc formate ([(CH3)2NH2]Zn(HCOO)3 or DMZnF) is a model system for the study of hybrid perovskite-like dielectrics. It undergoes a phase transition from the paraelectric to ferroelectric phase at ∼166 K, as observed via NMR spectra. The mechanism of this phase transition has been shown to have contributions from ordering of the hydrogen bonds between [(CH3)2NH2]+ (DMA+) and the formate groups as well as buckling of the metal-formate framework, but the transition dynamics and atomistic mechanism are not fully clear. This work presents dielectric constant measurements as evidence of cluster formation of the low-temperature phase and the relaxor-like behavior of this metal–organic framework above the phase transition temperature. 13C CP-MAS is used to track the evolution of the chemical shift, T1, and T2 of the dimethylammonium cation and formate groups from room temperature to 120 K. 2D 13C–13C correlation measurements provide evidence of the formation of pretransitional clusters above the phase transition temperature. Density functional theory (DFT) calculations support the assignment of chemical shifts and the proposed model. The analysis of 13C CP-MAS spectra and DFT calculations is used to discuss the mechanism of the dielectric phase transition and the origin of relaxor-like behavior in DMZnF.
Co-reporter:Jonathan H. Christian, David W. Brogden, Jasleen K. Bindra, Jared S. Kinyon, Johan van Tol, Jingfang Wang, John F. Berry, and Naresh S. Dalal
Inorganic Chemistry 2016 Volume 55(Issue 13) pp:6376
Publication Date(Web):February 16, 2016
DOI:10.1021/acs.inorgchem.5b02545
Magnetic properties of the series of three linear, trimetallic chain compounds Cr2Cr(dpa)4Cl2, 1, Mo2Cr(dpa)4Cl2, 2, and W2Cr(dpa)4Cl2, 3 (dpa = 2,2′-dipyridylamido), have been studied using variable-temperature dc and ac magnetometry and high-frequency EPR spectroscopy. All three compounds possess an S = 2 electronic ground state arising from the terminal Cr2+ ion, which exhibits slow magnetic relaxation under an applied magnetic field, as evidenced by ac magnetic susceptibility and magnetization measurements. The slow relaxation stems from the existence of an easy-axis magnetic anisotropy, which is bolstered by the axial symmetry of the compounds and has been quantified through rigorous high-frequency EPR measurements. The magnitude of D in these compounds increases when heavier ions are substituted into the trimetallic chain; thus D = −1.640, −2.187, and −3.617 cm–1 for Cr2Cr(dpa)4Cl2, Mo2Cr(dpa)4Cl2, and W2Cr(dpa)4Cl2, respectively. Additionally, the D value measured for W2Cr(dpa)4Cl2 is the largest yet reported for a high-spin Cr2+ system. While earlier studies have demonstrated that ligands containing heavy atoms can enhance magnetic anisotropy, this is the first report of this phenomenon using heavy metal atoms as “ligands”.
Co-reporter:Prinson P. Samuel, Roman Neufeld, Kartik Chandra Mondal, Herbert W. Roesky, Regine Herbst-Irmer, Dietmar Stalke, Serhiy Demeshko, Franc Meyer, Vallyanga Chalil Rojisha, Susmita De, Pattiyil Parameswaran, A. Claudia Stückl, Wolfgang Kaim, Jonathan H. Christian, Jasleen K. Bindra and Naresh S. Dalal
Chemical Science 2015 vol. 6(Issue 5) pp:3148-3153
Publication Date(Web):20 Mar 2015
DOI:10.1039/C5SC00646E
Cr(I)Cl is a very unstable species. The present work describes the stabilisation of Cr(I)Cl in the low coordinate environment of cyclic alkyl(amino) carbene ligands and its synthetic application to yield an unprecedented cationic complex with a two coordinate Cr(I). One electron reduction of (cAAC)2CrCl2 (1) with equivalent amount of KC8 results in the formation of (cAAC)2CrCl (2), with a distorted trigonal planar configuration at the metal centre. SQUID, EPR and theoretical studies reveal a Cr(I) centre with S = 5/2 spin ground state for 2. It represents the first example of a mononuclear Cr complex showing slow relaxation of magnetisation under an applied magnetic field. The chlorine atom in 2 is expected to be prone to further reactions with appropriate reagents. This qualifies 2 as a promising precursor for the preparation of various interesting complexes with Cr(I) in a low coordinate environment. The first example of this metathesis reaction is observed when 2 is treated with Na[B(C6H3(CF3)2)4] resulting in [(cAAC)2Cr]+[B(C6H3(CF3)2)4]−, a linear cationic complex with two coordinate Cr(I) and an S = 5/2 spin ground state.
Co-reporter:Vasanth Ramachandran, Johan van Tol, Amy M. McKenna, Ryan P. Rodgers, Alan G. Marshall, and Naresh S. Dalal
Analytical Chemistry 2015 Volume 87(Issue 4) pp:2306
Publication Date(Web):February 3, 2015
DOI:10.1021/ac504080g
In the first use of high-field electron paramagnetic resonance (EPR) spectroscopy to characterize paramagnetic metal–organic and free radical species from tar balls and weathered crude oil samples from the Gulf of Mexico (collected after the Deepwater Horizon oil spill) and an asphalt volcano sample collected off the coast of Santa Barbara, CA, we are able to identify for the first time the various paramagnetic species present in the native state of these samples and understand their molecular structures and bonding. The two tar ball and one asphalt volcano samples contain three distinct paramagnetic species: (i) an organic free radical, (ii) a [VO]2+ containing porphyrin, and (iii) a Mn2+ containing complex. The organic free radical was found to have a disc-shaped or flat structure, based on its axially symmetric spectrum. The characteristic spectral features of the vanadyl species closely resemble those of pure vanadyl porphyrin; hence, its nuclear framework around the vanadyl ion must be similar to that of vanadyl octaethyl porphyrin (VOOEP). The Mn2+ ion, essentially undetected by low-field EPR, yields a high-field EPR spectrum with well-resolved hyperfine features devoid of zero-field splitting, characteristic of tetrahedral or octahedral Mn–O bonding. Although the lower-field EPR signals from the organic free radicals in fossil fuel samples have been investigated over the last 5 decades, the observed signal was featureless. In contrast, high-field EPR (up to 240 GHz) reveals that the species is a disc-shaped hydrocarbon molecule in which the unpaired electron is extensively delocalized. We envisage that the measured g-value components will serve as a sensitive basis for electronic structure calculations. High-field electron nuclear double resonance experiments should provide an accurate picture of the spin density distribution for both the vanadyl-porphyrin and Mn2+ complexes, as well as the organic free radical, and will be the focus of follow-up studies.
Structure–Property Correlations in the Heterobimetallic 4f/3d Materials Ln2M(TeO3)2(SO4) (Ln = Y, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, or Lu; M = Co or Zn)
Co-reporter:Jian Lin, Kariem Diefenbach, Mark A. Silver, Naresh S. Dalal, and Thomas E. Albrecht-Schmitt
Crystal Growth & Design 2015 Volume 15(Issue 9) pp:4606-4615
Publication Date(Web):August 13, 2015
DOI:10.1021/acs.cgd.5b00860
Eighteen new lanthanide transition metal heterobimetallic compounds, Ln2Co(TeO3)2(SO4)2 (Ln = Y, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, or Lu) and Ln2Zn(TeO3)2(SO4)2 (Ln = Sm, Gd, Dy, Ho, Er, or Yb), have been prepared. They crystallize in triclinic space group P1̅ with two different structural topologies occurring because of a reduction in the Ln3+ coordination number from eight to seven with the smallest lanthanides, Yb3+ and Lu3+. Magnetic susceptibility studies of compounds with diamagnetic lanthanides and lanthanide-like ions suggest that antiferromagnetic interactions occur between the Co2+ ions. Similarly, the replacement of Co2+ with Zn2+ yields Ln2Zn(TeO3)2(SO4)2 (Ln = Gd, Dy, Ho, or Er), and these materials allow for the resolution of the nature of the interactions between lanthanide ions. The data suggest that the short-range Ln3+···Ln3+ interactions are ferromagnetic. However, a wide range of ferro- and antiferromagnetic interactions occur between the Ln3+ and the Co2+ cations, with several compounds exhibiting short-range magnetic correlations below 25 K. The results are discussed and contrasted with those recently reported for the related Ln2Cu(TeO3)2(SO4)2 family.
Co-reporter:Nan Xu, Jonathan H. Christian, Naresh S. Dalal, Erwin G. Abucayon, Colin Lingafelt, Douglas R. Powell and George B. Richter-Addo
Dalton Transactions 2015 vol. 44(Issue 46) pp:20121-20130
Publication Date(Web):28 Oct 2015
DOI:10.1039/C5DT03074A
NONOates (diazeniumdiolates) containing the [X{N2O2}]− functional group are frequently employed as nitric oxide (NO) donors in biology, and some NONOates have been shown to bind to metalloenzymes. We report the preparation, crystal structures, detailed magnetic behavior, redox properties, and reactivities of the first isolable alkyl C-NONOate complexes of heme models, namely (OEP)Fe(η2-ON(t-Bu)NO) (1) and (TPP)Fe(η2-ON(t-Bu)NO) (2) (OEP = octaethylporphyrinato dianion, TPP = tetraphenylporphyrinato dianion). The compounds display the unusual NONOate O,O-bidentate binding mode for porphyrins, resulting in significant apical Fe displacements (+0.60 Å for 1, and +0.69 Å for 2) towards the axial ligands. Magnetic susceptibility and magnetization measurements made from 1.8–300 K at magnetic fields from 0.02 to 5 T, yielded magnetic moments of 5.976 and 5.974 Bohr magnetons for 1 and 2, respectively, clearly identifying them as high-spin (S = 5/2) ferric compounds. Variable-frequency (9.4 GHz and 34.5 GHz) EPR measurements, coupled with computer simulations, confirmed the magnetization results and yielded more precise values for the spin Hamiltonian parameters: gavg = 2.00 ± 0.03, |D| = 3.89 ± 0.09 cm−1, and E/D = 0.07 ± 0.01 for both compounds, where D and E are the axial and rhombic zero-field splittings. IR spectroelectrochemistry studies reveal that the first oxidations of these compounds occur at the porphyrin macrocycles and not at the Fe-NONOate moieties. Reactions of 1 and 2 with a histidine mimic (1-methylimidazole) generate RNO and NO, both of which may bind to the metal center if sterics allow, as shown by a comparative study with the Cupferron complex (T(p-OMe)PP)Fe(η2-ON(Ph)NO). Protonation of 1 and 2 yields N2O as a gaseous product, presumably from the initial generation of HNO that dimerizes to the observed N2O product.
Co-reporter:Jin Jung Kweon
The Journal of Physical Chemistry C 2015 Volume 119(Issue 9) pp:5013-5019
Publication Date(Web):February 20, 2015
DOI:10.1021/jp512127x
Co-reporter:Nandita Abhyankar
The Journal of Physical Chemistry C 2015 Volume 119(Issue 50) pp:28143-28147
Publication Date(Web):November 23, 2015
DOI:10.1021/acs.jpcc.5b10326
We employ electron paramagnetic resonance (EPR) of Mn2+ as a spin probe to study the paraelectric–ferroelectric transition in dimethylammonium manganese formate, [(CH3)2NH2]Mn(CHCO2)3, (DMMnF), which is considered a model metal–organic framework (MOF) with a Pb-free perovskite architecture. We study the variation of the Mn2+ EPR line shape and intensity at the X-band (∼9.5 GHz) over 80 to 300 K. The peaks are essentially Lorentzian, implying electron spin exchange at frequencies greater than 9.5 GHz. On cooling, an anomalous increase in the peak width is noted at 185 K but no anomalous change in the normalized, double-integrated EPR signal intensity around the TC, indicating that DMMnF is transparent to microwave electric fields with a clear lack of magnetoelectric coupling, in contrast to an earlier report. Our analysis enables us to estimate change in lattice strain related to the ferroelectric transition, information that is difficult to obtain by other techniques.
Co-reporter:Prinson P. Samuel ; Kartik Chandra Mondal ; Nurul Amin Sk ; Herbert W. Roesky ; Elena Carl ; Roman Neufeld ; Dietmar Stalke ; Serhiy Demeshko ; Franc Meyer ; Liviu Ungur ; Liviu F. Chibotaru ; Jonathan Christian ; Vasanth Ramachandran ; Johan van Tol
Journal of the American Chemical Society 2014 Volume 136(Issue 34) pp:11964-11971
Publication Date(Web):July 29, 2014
DOI:10.1021/ja5043116
Cyclic alkyl(amino) carbene stabilized two- and three-coordinate Fe(I) complexes, (cAAC)2FeCl (2) and [(cAAC)2Fe][B(C6F5)4] (3), respectively, were prepared and thoroughly studied by a bouquet of analytical techniques as well as theoretical calculations. Magnetic susceptibility and Mössbauer spectroscopy reveal the +1 oxidation state and S = 3/2 spin ground state of iron in both compounds. 2 and 3 show slow magnetic relaxation typical for single molecule magnets under an applied direct current magnetic field. The high-frequency EPR measurements confirm the S = 3/2 ground state with a large, positive zero-field splitting (∼20.4 cm–1) and reveal easy plane anisotropy for compound 2. CASSCF/CASPT2/RASSI-SO ab initio calculations using the MOLCAS program package support the experimental results.
Co-reporter:Wenjing Liu, Jonathan H. Christian, Rami Al-Oweini, Bassem S. Bassil, Johan van Tol, Mihail Atanasov, Frank Neese, Naresh S. Dalal, and Ulrich Kortz
Inorganic Chemistry 2014 Volume 53(Issue 17) pp:9274-9283
Publication Date(Web):August 19, 2014
DOI:10.1021/ic501385r
Two monochromium(III)-containing heteropolytungstates, [CrIII(HPVW7O28)2]13- (1a) and [CrIII(HAsVW7O28)2]13- (2a), were prepared via simple, one-pot reactions in aqueous, basic medium, by reaction of the composing elements, and then isolated as hydrated sodium salts, Na13[CrIII(HPVW7O28)2]·47H2O (1) and Na13[CrIII(HAsVW7O28)2]·52H2O (2). Polyanions 1a and 2a comprise an octahedrally coordinated CrIII ion, sandwiched by two {PW7} or {AsW7} units. Both compounds 1 and 2 were fully characterized in the solid state by single-crystal XRD, IR spectroscopy, thermogravimetric and elemental analyses, magnetic susceptibility, and EPR measurements. Magnetic studies on 1 and 2 demonstrated that both compounds exhibit appreciable deviation from typical paramagnetic behavior, and have a ground state S = 3/2, as expected for a CrIII ion, but with an exceptionally large zero-field uniaxial anisotropy parameter (D). EPR measurements on powder and single-crystal samples of 1 and 2 using 9.5, 34.5, and 239.2 GHz frequencies and over 4–295 K temperature fully support the magnetization results and show that D = +2.4 cm–1, the largest and sign-assigned D-value so far reported for an octahedral CrIII-containing, molecular compound. Ligand field analysis of results from CASSCF and NEVPT2-correlated electronic structure calculations on Cr(OH)63– model complexes allowed to unravel the crucial role of the second coordination sphere of CrIII for the unusually large magnetic anisotropy reflected by the experimental value of D. The newly developed theoretical modeling, combined with the synthetic procedure for producing such unusual magnetic molecules in a well-defined and essentially magnetically isolated environment, appears to be a versatile new research area.
Co-reporter:Jin Jung Kweon ; Riqiang Fu ; Eden Steven ; Cheol Eui Lee
The Journal of Physical Chemistry C 2014 Volume 118(Issue 25) pp:13387-13393
Publication Date(Web):June 13, 2014
DOI:10.1021/jp501531h
LiH2PO4 (LDP) is a favored candidate for hydrogen fuel cells, but the mechanism of its high protonic conductivity remains unclear. A complicating factor has been the lack of resolution in the reported proton NMR spectra. We now report multinuclear magic angle spinning NMR in LDP at magnetic fields up to 21.2 T. Well-resolved 1H NMR spectra are observed that are assignable to protons in the short and long O–H···O hydrogen bonds and a peak to physisorbed H2O. The position and intensity for the H2O peak depend on the H2O content, implying fast exchange between the adsorbed H2O and the O–H···O protons. 31P and 7Li NMR spectra and spin–lattice relaxation measurements showed that the proton hopping/exchange processes involve concerted hindered rotational fluctuations of the phosphate groups. Conductivity data from adsorbed H2O-controlled samples clearly suggest that the mechanism of LDP’s protonic conductivity is dominantly the exchange (and hopping) of the adsorbed H2O protons with the short O–H···O hydrogen bonds, in contrast to an earlier model that ascribed it to intermolecular hopping of O–H···O protons. The new findings enable us to modulate LDP’s protonic conductivity by several orders of magnitude via controlling physisorbed water.
Co-reporter:Lakshmi Kanta Das ; Apurba Biswas ; Jared S. Kinyon ; Naresh S. Dalal ; Haidong Zhou ;Ashutosh Ghosh
Inorganic Chemistry 2013 Volume 52(Issue 20) pp:11744-11757
Publication Date(Web):October 3, 2013
DOI:10.1021/ic401020m
Oxime-based tridentate Schiff base ligands 3-[2-(diethylamino)ethylimino]butan-2-one oxime (HL1) and 3-[3-(dimethylamino)propylimino]butan-2-one oxime (HL2) produced the dinuclear complex [Ni2L12](ClO4)2 (1) and trinuclear complex [Ni3(HL2)3(μ3-O)](ClO4)4·CH3CN (2), respectively, upon reaction with Ni(ClO4)2·6H2O. However, in a slightly alkaline medium, both of the ligands underwent hydrolysis and resulted in tetranuclear complexes [{Ni(deen)(H2O)}2(μ3-OH)2{Ni2(moda)4}](ClO4)2·2CH3CN (3) and [{Ni(dmpn)(CH3CN)2}2(μ3-OH)2{Ni2(moda)4}](ClO4)2·CH3CN (4), where deen = 2-(diethylamino)ethylamine, dmpn = 3-(dimethylamino)-1-propylamine, and modaH = diacetyl monoxime. All four complexes have been structurally characterized. Complex 1 is a centrosymmetric dimer where the square planar nickel(II) atoms are joined solely by the oximato bridges. In complex 2, three square planar nickel atoms form a triangular core through a central oxido (μ3-O) and peripheral oximato bridges. Tetranuclear complexes 3 and 4 consist of four distorted octahedral nickel(II) ions held together in a rhombic chair arrangement by two central μ3-OH and four peripheral oximato bridges. Magnetic susceptibility measurements indicated that dinuclear 1 and trinuclear 2 exhibited diamagnetic behavior, while tetranuclear complexes 3 and 4 were found to have dominant antiferromagnetic intramolecular coupling with concomitant ferromagnetic interactions. Despite its singlet ground state, both 3 and 4 serve as useful examples of Kahn’s model for competing spin interactions. High-frequency EPR studies were also attempted, but no signal was detected, likely due to the large energy gap between the ground and first excited state. Complexes 3 and 4 exhibited excellent catecholase-like activity in the aerial oxidation of 3,5-di-tert-butylcatechol to the corresponding o-quinone, whereas 1 and 2 did not show such catalytic activity. Kinetic data analyses of this oxidation reaction in acetonitrile revealed that the catalytic activity of 3 (kcat = 278.4 h–1) was slightly lower than that of 4 (kcat = 300.0 h–1). X-band EPR spectroscopy indicated that the reaction proceeded through the formation of iminoxyl-type radicals.
Co-reporter:Raghabendra Samantaray ; Ronald J. Clark ; Eun S. Choi
Journal of the American Chemical Society 2012 Volume 134(Issue 38) pp:15953-15962
Publication Date(Web):August 30, 2012
DOI:10.1021/ja3065705
The antiferromagnetic Cr(V) peroxychromates, M3Cr(O2)4, M = K, Rb, and Cs, become ferroelectric when mixed with NH4+, but the underlying mechanism is not understood. Our dielectric relaxation, Raman scattering, and high-frequency EPR measurements on the M3–x(NH4)xCr(O2)4 family clarify this mechanism. At 295 K, (NH4)3Cr(O2)4 is tetragonal (I4̅2m), with the NH4+ ions occupying two distinctly different sites, N1 and N2. A ferroelectric transition at Tc1 = 250 K is revealed by λ-type anomalies in Cp and dielectric constant, and lowering of symmetry to Cmc2(1). Below Tc1, the N1 sites lose their tetrahedral symmetry and thus polarization develops. Raman detection of translational modes involving the NH4+ ions around 193 cm–1 supports this model. EPR around Tc1 revealed that the [Cr(O2)4]3– ions reorient by about 10°. A minor peak at Tc2 ≈ 207 K is attributed to a short-range ordering that culminates in a long-range, structural order at Tc3 ≈ 137 K. At Tc3, the symmetry is lowered to P1 with significant changes in the cell parameters. Rb+ and Cs+ substitutions that block the N1 and N2 sites selectively show that Tc1 is related to the torsional motion of the N1 site, while Tc2 and Tc3 are governed by the motional slowing down of the N2 site. These data show that the multiferroic behavior of this family is governed by the rotational and translational dynamics of the NH4+ ions and is tunable by their controlled substitutions. Relevance to other classes of possible multiferroics is pointed out.
Co-reporter:Pampa M. Guha, Hoa Phan, Jared S. Kinyon, Wendy S. Brotherton, Kesavapillai Sreenath, J. Tyler Simmons, Zhenxing Wang, Ronald J. Clark, Naresh S. Dalal, Michael Shatruk, and Lei Zhu
Inorganic Chemistry 2012 Volume 51(Issue 6) pp:3465-3477
Publication Date(Web):March 7, 2012
DOI:10.1021/ic2021319
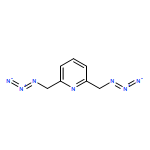
Copper(II) acetate mediated coupling reactions between 2,6-bis(azidomethyl)pyridine or 2-picolylazide and two terminal alkynes afford 1,2,3-triazolyl-containing ligands L1–L6. These ligands contain various nitrogen-based Lewis basic sites including two different pyridyls, two nitrogen atoms on a 1,2,3-triazolyl ring, and the azido group. A rich structural diversity, which includes mononuclear and dinuclear complexes as well as one-dimensional polymers, was observed in the copper(II) complexes of L1–L6. The preference of copper(II) to two common bidentate 1,2,3-triazolyl-containing coordination sites was investigated using isothermal titration calorimetry and, using zinc(II) as a surrogate, in 1H NMR titration experiments. The magnetic interactions between the copper(II) centers in three dinuclear complexes were analyzed via temperature-dependent magnetic susceptibility measurements and high-frequency electron paramagnetic resonance spectroscopy. The observed magnetic superexchange is strongly dependent on the orientation of magnetic orbitals of the copper(II) ions and can be completely turned off if these orbitals are arranged orthogonal to each other. This work demonstrates the versatility of 1,2,3-triazolyl-containing polyaza ligands in forming metal coordination complexes of a rich structural diversity and interesting magnetic properties.
Co-reporter:Gina M. Chiarella ; F. Albert Cotton ; Naresh S. Dalal ; Carlos A. Murillo ; Zhenxing Wang ;Mark D. Young
Inorganic Chemistry 2012 Volume 51(Issue 9) pp:5257-5263
Publication Date(Web):April 16, 2012
DOI:10.1021/ic300169f
Three rare compounds have been synthesized and structurally characterized; these species have paddlewheel structures and Re27+ cores surrounded by four bicyclic guanidinates and two axial ligands along the Re–Re axis. Each possesses a formal bond order of 3.5 and a σ2π4δ1 electronic configuration that entails the presence of one unpaired electron for each compound. The guanidinate ligands characterized by having CH2 entities and a central C(N)3 unit that joins two cyclic units—one having two fused 6-membered rings (hpp) and the other having a 5- and a 6-membered ring fused together (tbn)—allowed the isolation of [Re2(tbn)4Cl2]PF6, 1, [Re2(tbn)4Cl2]Cl, 2, and [Re2(hpp)4(O3SCF3)2](O3SCF3), 3. Because of the larger bite angle of the tbn relative to the hpp ligand, the Re–Re bond distances in 1 and 2 (2.2691(14) and 2.2589(14) Å, respectively) are much longer than that in 3 (2.1804(8) Å). Importantly, electron paramagnetic resonance (EPR) studies at both X-band (∼9.4 GHz) and W-band (112 GHz) in the solid and in frozen solution show unusually low g-values (∼1.75) and the absence of zero-field splitting, providing direct evidence for the presence of one metal-based unpaired electron for both 1 and 3. These spectroscopic data suggest that the unsymmetrical 5-/6-membered ligand leads to the formation of isomers, as shown by significantly broader EPR signals for 1 than for 3, even though both compounds possess what appears to be similar ideal crystallographic axial symmetry on the X-ray time scale.
Polyoxopalladates Encapsulating 8-Coordinated Metal Ions, [MO8PdII12L8]n− (M = Sc3+, Mn2+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Lu3+; L = PhAsO32–, PhPO32–, SeO32–)
Co-reporter:Maria Barsukova-Stuckart, Natalya V. Izarova, Ryan A. Barrett, Zhenxing Wang, Johan van Tol, Harold W. Kroto, Naresh S. Dalal, Pablo Jiménez-Lozano, Jorge J. Carbó, Josep M. Poblet, Marc S. von Gernler, Thomas Drewello, Pedro de Oliveira, Bineta Keita, and Ulrich Kortz
Inorganic Chemistry 2012 Volume 51(Issue 24) pp:13214-13228
Publication Date(Web):November 29, 2012
DOI:10.1021/ic301537n
A total of 16 discrete polyoxopalladates(II) [MO8PdII12L8]n−, with a metal ion M encapsulated in a cuboid-shaped {Pd12O8L8} cage, have been synthesized: the phenylarsonate-capped series (1) L = PhAsO32–, M = Sc3+ (ScPhAs), Mn2+ (MnPhAs), Fe3+ (FePhAs), Co2+ (CoPhAs), Ni2+ (NiPhAs), Cu2+ (CuPhAs), Zn2+ (ZnPhAs); the phenylphosphonate-capped series: (2) L = PhPO32–, M = Cu2+ (CuPhP), Zn2+ (ZnPhP); and the selenite-capped series (3) L = SeO32–, M = Mn2+ (MnSe), Fe3+ (FeSe), Co2+ (CoSe), Ni2+ (NiSe), Cu2+, (CuSe), Zn2+ (ZnSe), Lu3+ (LuSe)). The polyanions were prepared in one-pot reactions in aqueous solution of [Pd3(CH3COO)6] with an appropriate salt of the metal ion M, as well as PhAsO3H2, PhPO3H2, and SeO2, respectively, and then isolated as hydrated sodium salts Nan[MO8PdII12L8]·yH2O (y = 10–37). The compounds were characterized in the solid state by IR spectroscopy, single-crystal XRD, elemental and thermogravimetric analyses. The solution stability of the diamagnetic polyanions ScPhAs, ZnPhAs, ZnPhP, ZnSe, and LuSe was confirmed by multinuclear (77Se, 31P, 13C, and 1H) NMR spectroscopy. The polyoxopalladates ScPhAs, MnPhAs, CoPhAs, and CuPhAs were investigated by electrospray ionization mass spectrometry (ESI-MS) and tandem mass spectrometry (MS/MS). Electrochemical studies on the manganese- and iron-containing derivatives demonstrated that the redox properties of the Mn2+, Fe3+, and Pd2+ centers in the polyanions are strikingly influenced by the nature of the capping group. These results have subsequently been verified by density functional theory (DFT) calculations. Interestingly, electron paramagnetic resonance (EPR) measurements suggest that the coordination geometry around Mn2+ is dynamically distorted on the EPR time scale (∼10–11 s), whereas it appears as a static ensemble with cubic symmetry on the X-ray diffraction (XRD) time-scale (10–15 s). The octacoordinated Cu2+ cuboid is similarly distorted, in good agreement with DFT calculations. Interestingly, g∥ is smaller than g⊥, which is quite unusual, needing further theoretical development.
Co-reporter:Zhenxing Wang, Weiwei Zheng, Johan van Tol, Naresh S. Dalal, Geoffrey F. Strouse
Chemical Physics Letters 2012 Volume 524() pp:73-77
Publication Date(Web):6 February 2012
DOI:10.1016/j.cplett.2011.12.038
Abstract
High-frequency electron paramagnetic resonance (HF-EPR) provides a contactless microscopic probe of magnetic impurities and their surroundings in nanoparticles. In 5.0 nm colloidally prepared CdSe quantum dots (QDs) containing 0.6% Mn2+, a core and a surface site can be readily distinguished on the basis of g-factor and 55Mn hyperfine interaction. In contrast to EPR in the bulk, at low temperatures a broad background signal develops, which at high fields is the only remaining feature. Previous studies have suggested this background signal originates from exchange coupled Mn2+ clusters arising from spinodal decomposition; HF-EPR shows that it is due to strain-broadening of the zero-field splitting related to local lattice distortions within the QD.
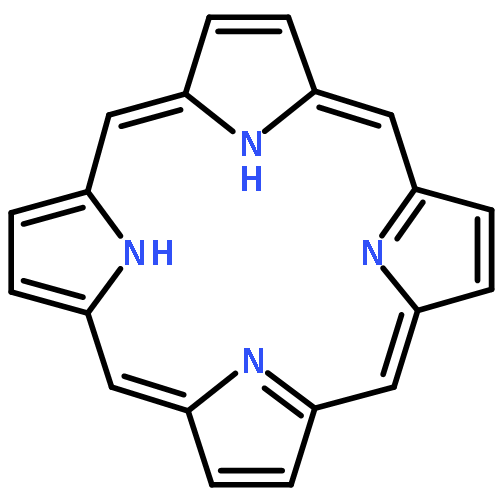
Co-reporter:Sneha Dugar, Riqiang Fu, and Naresh S. Dalal
The Journal of Physical Chemistry B 2012 Volume 116(Issue 30) pp:9215-9222
Publication Date(Web):June 8, 2012
DOI:10.1021/jp302189r
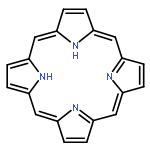
Octaethyl porphyrin (OEP) and its Ni and Zn derivatives are considered as model compounds in biochemical, photophysical, and fossil fuel chemistry. They have thus been investigated by high-resolution solid-state 13C NMR using powders, but peak assignment has been difficult because of large line widths. Arguing that a significant cause of broadening might be the anisotropic bulk magnetic susceptibility, we utilized single crystals in our 13C cross-polarization magic angle spinning (CP-MAS) measurements and observed a nearly 2-fold line narrowing. This enhanced resolution enabled us to assign chemical shifts to each carbon for all the three compounds. The new assignments are now in agreement with X-ray structural data and allowed us to probe the motional dynamics of the methyl and methylene carbons of the OEP side chains. It is apparent that the use of single crystals in 13C CP-MAS measurements has a significantly wider impact than previously thought.
Co-reporter:Raghabendra Samantaray ; Ronald J. Clark ; Eun S. Choi ; Haidong Zhou
Journal of the American Chemical Society 2011 Volume 133(Issue 11) pp:3792-3795
Publication Date(Web):February 28, 2011
DOI:10.1021/ja1117683
Upon consideration of the hydrogen-bonding properties of the NH4+ cation, we synthesized a new class of compounds, M3−x(NH4)xCrO8 (M = Na, K, Rb, Cs). These magnetic compounds with the simple 3d1 ground state become ferroelectric. X-ray studies confirmed that the phase transition involves a symmetry change from I4̅2m to Cmc21 to P1. The transition temperature depends linearly on the composition variable x. The transitions are of the order−disorder type, with N−H···O bonding playing the central role in the mechanism. Extension of this idea to the introduction of ferroelectricity in several other classes of materials is suggested.
Co-reporter:Zhenxing Wang ; Johan van Tol ; Taketo Taguchi ; Matthew R. Daniels ; George Christou
Journal of the American Chemical Society 2011 Volume 133(Issue 44) pp:17586-17589
Publication Date(Web):October 9, 2011
DOI:10.1021/ja207636b
A high spin (S) compound has been synthesized whose properties straddle the interface between the classical and quantum mechanical spin descriptions. The cluster [Mn7O4(pdpm)6(N3)4](ClO4)2 (Mn7) has an unprecedented core structure comprising an octahedral [MnIII6(μ4-O)(μ3-O)3(μ3-N3)4]6+ unit with one of its faces capped by a MnII ion. Magnetization and susceptibility studies indicate an S = 29/2 ground state, the maximum possible. Variable-temperature, high-frequency electron paramagnetic resonance (HF-EPR) spectra on powder and single-crystal samples of Mn7 exhibit sharp spectral features characteristic of a quantum spin that are well resolved in a certain temperature range but which transform to a continuum of peaks characteristic of a classical spin in another; these features have been well reproduced by computer simulations. A fast Fourier transform analysis of the sharp spectral features and the low temperature EPR spectra suggests that more than one spin state are involved.
Co-reporter:Maria Barsukova-Stuckart;Dr. Natalya V. Izarova; Geoffrey B. Jameson;Vasanth Ramachran;Zhenxing Wang;Dr. Johan vanTol; Naresh S. Dalal;Rosa NgoBiboum;Dr. Bineta Keita; Louis Nadjo; Ulrich Kortz
Angewandte Chemie International Edition 2011 Volume 50( Issue 11) pp:2639-2642
Publication Date(Web):
DOI:10.1002/anie.201006734
Co-reporter:Tiglet Besara;Prashant Jain;Philip L. Kuhns;Arneil P. Reyes;Harold W. Kroto;Anthony K. Cheetham
PNAS 2011 Volume 108 (Issue 17 ) pp:6828-6832
Publication Date(Web):2011-04-26
DOI:10.1073/pnas.1102079108
Transitions associated with orientational order–disorder phenomena are found in a wide range of materials and may have a significant
impact on their properties. In this work, specific heat and 1H NMR measurements have been used to study the phase transition in the metal-organic framework (MOF) compound [(CH3)2NH2]Zn(HCOO)3. This compound, which possesses a perovskite-type architecture, undergoes a remarkable order–disorder phase transition at
156 K. The (DMA+) cationic moieties that are bound by hydrogen bonds to the oxygens of the formate groups (N─H⋯O ∼ 2.9 Å) are essentially trapped inside the basic perovskite cage architecture. Above 156 K, it is the orientations of these moieties
that are responsible for the disorder, as each can take up three different orientations with equal probability. Below 156 K,
the DMA+ is ordered within one of these sites, although the moiety still retains a considerable state of motion. Below 40 K, the rotational
motions of the methyl groups start to freeze. As the temperature is increased from 4 K in the NMR measurements, different
relaxation pathways can be observed in the temperature range approximately 65–150 K, as a result of a “memory effect.” This
dynamic behavior is characteristic of a glass in which multiple states possess similar energies, found here for a MOF. This
conclusion is strongly supported by the specific heat data.
Co-reporter:Jingfang Wang, Zhenxing Wang, Ronald J. Clark, Andrew Ozarowski, Johan van Tol, Naresh S. Dalal
Polyhedron 2011 30(18) pp: 3058-3061
Publication Date(Web):
DOI:10.1016/j.poly.2011.02.032
Co-reporter:Dr. Bassem S. Bassil;Masooma Ibrahim;Rami Al-Oweini;Marie Asano;Zhenxing Wang;Dr. Johan vanTol; Naresh S. Dalal; Kwang-Yong Choi;Rosa NgoBiboum;Dr. Bineta Keita; Louis Nadjo; Ulrich Kortz
Angewandte Chemie International Edition 2011 Volume 50( Issue 26) pp:5961-5964
Publication Date(Web):
DOI:10.1002/anie.201007617
Co-reporter:Maria Barsukova-Stuckart;Dr. Natalya V. Izarova; Geoffrey B. Jameson;Vasanth Ramachran;Zhenxing Wang;Dr. Johan vanTol; Naresh S. Dalal;Rosa NgoBiboum;Dr. Bineta Keita; Louis Nadjo; Ulrich Kortz
Angewandte Chemie 2011 Volume 123( Issue 11) pp:2688-2692
Publication Date(Web):
DOI:10.1002/ange.201006734
Co-reporter:Dr. Bassem S. Bassil;Masooma Ibrahim;Rami Al-Oweini;Marie Asano;Zhenxing Wang;Dr. Johan vanTol; Naresh S. Dalal; Kwang-Yong Choi;Rosa NgoBiboum;Dr. Bineta Keita; Louis Nadjo; Ulrich Kortz
Angewandte Chemie 2011 Volume 123( Issue 26) pp:6083-6087
Publication Date(Web):
DOI:10.1002/ange.201007617
Co-reporter:Michael Nippe ; Jingfang Wang ; Eckhard Bill ; Håkon Hope ; Naresh S. Dalal ;John F. Berry
Journal of the American Chemical Society 2010 Volume 132(Issue 40) pp:14261-14272
Publication Date(Web):September 22, 2010
DOI:10.1021/ja106510g
Crystal structures of the heterometallic compounds CrCrFe(dpa)4Cl2 (1), CrCrMn(dpa)4Cl2 (2), and MoMoMn(dpa)4Cl2 (3) (dpa = 2,2′-dipyridylamide) show disorder in the metal atom positions such that the linear MAMA···MB array for a given molecule in the crystal is oriented in one of two opposing directions. Despite the fact that the direct coordination sphere of the metals in the two crystallographically independent orientations is identical, subtle differences in some metal−ligand bond distances are observed in 1 and 3 due to differences in the orientation of a solvent molecule of crystallization. The Fe(II) and Mn(II) ions serve as sensitive local spectroscopic probes that have been interrogated by Mössbauer spectroscopy and high-field EPR spectroscopy, respectively. The subtle differences in the two independent Fe and Mn sites in 1 and 3 unexpectedly give rise to unusually large differences in the measured Fe quadrupole splitting (ΔEQ) in 1 and Mn zero-field splitting (D) in 3. Variable-temperature/single-crystal EPR spectroscopy has allowed us to determine that the temperature-dependent D tensors in 3 are oriented along the metal−metal axis and that they show significantly different dynamic behavior with temperature. The differences in ΔEQ and D are reproduced by density functional calculations on truncated models for 1 and 3 that lack the quadruply bonded MAMA groups, though the magnitude of the calculated effect is not as large as that observed experimentally. We suggest that the large observed differences in ΔEQ and D for the individual sites could be due to the influence of the strong diamagnetic anisotropy of the quadruply bonded MM unit.
Co-reporter:F. Albert Cotton;Narpinder Kaur
Journal of Cluster Science 2010 Volume 21( Issue 3) pp:301-312
Publication Date(Web):2010 September
DOI:10.1007/s10876-010-0289-7
Oxidation of α-[Mo2(cis-DAniF)2]3(μ-F)6 with an excess of FeCp2BF4 produces the triply oxidized species {β-[Mo2(cis-DAniF)2]3(μ-F)6}(BF4)3. During the oxidation process, the conformation in the α triangular species changes from an arrangement in which two dimetal units are parallel and the third one essentially orthogonal to a structure in which all three dimetal units are parallel. Furthermore, upon removal of three electrons, the Mo–Mo distances increase by about 0.05–0.06 Å and the Mo–F bond distances decrease by 0.04 Å. The structural data, as well as EPR, are consistent with an electronically localized system and a decrease in bond order from 4 to 3.5 for each dimetal unit.
Co-reporter:R.M Achey, P.L Kuhns, A.P Reyes, W.G Moulton, N.S Dalal
Solid State Communications 2002 Volume 121(2–3) pp:107-109
Publication Date(Web):2 January 2002
DOI:10.1016/S0038-1098(01)00463-X
13C NMR measurements have been carried out on 13CH3-labeled Mn12O12-acetate in both powder and magnetically aligned samples at an applied magnetic field of 8.5 T. The four observed peaks are assignable to the four distinct 13CH3 moieties. The spectra were measured as a function of the orientation of the Zeeman field with respect to the easy axis in an aligned powder sample. The line positions were fitted to the dipolar plus isotropic interactions and the Fermi-contact parts, and unpaired electron spin density was calculated for each site. The results demonstrate significant spin density delocalization onto the acetate fragments and complement recent proton NMR and polarized neutron diffraction data on this system.
Co-reporter:Brant Cage, Philan Nguyen, Naresh Dalal
Solid State Communications 2001 Volume 119(10–11) pp:597-601
Publication Date(Web):29 August 2001
DOI:10.1016/S0038-1098(01)00296-4
This study presents the first observation of long-range antiferromagnetic order in a simple Cr(IV)-peroxo, S=1 complex. Magnetic susceptibility, χ, and heat capacity, Cp, results are presented for both powders and single crystals of triamminodiperoxychromate, Cr(NH3)3(O2)2. For the investigated compound, the spin state of S=1 was determined by analysis of χ from 2 to 200 K, and correlated with the entropy expected for such a system as determined by Cp measurements from 2.5 to 20 K. Sharp λ-like transitions were observed in both χ and Cp, with maxima at 8.80 and 8.46 K, respectively; characteristic of three-dimensional long-range antiferromagnetic ordering. The χ and Cp results correlated well using the Fisher heat capacity relationship. Further analysis of Cp determined that the majority of the spin entropy is engaged at temperatures below 8.46 K. This class of compounds might serve as precursors for CrO2 related materials.
Co-reporter:Nan Xu, Jonathan H. Christian, Naresh S. Dalal, Erwin G. Abucayon, Colin Lingafelt, Douglas R. Powell and George B. Richter-Addo
Dalton Transactions 2015 - vol. 44(Issue 46) pp:NaN20130-20130
Publication Date(Web):2015/10/28
DOI:10.1039/C5DT03074A
NONOates (diazeniumdiolates) containing the [X{N2O2}]− functional group are frequently employed as nitric oxide (NO) donors in biology, and some NONOates have been shown to bind to metalloenzymes. We report the preparation, crystal structures, detailed magnetic behavior, redox properties, and reactivities of the first isolable alkyl C-NONOate complexes of heme models, namely (OEP)Fe(η2-ON(t-Bu)NO) (1) and (TPP)Fe(η2-ON(t-Bu)NO) (2) (OEP = octaethylporphyrinato dianion, TPP = tetraphenylporphyrinato dianion). The compounds display the unusual NONOate O,O-bidentate binding mode for porphyrins, resulting in significant apical Fe displacements (+0.60 Å for 1, and +0.69 Å for 2) towards the axial ligands. Magnetic susceptibility and magnetization measurements made from 1.8–300 K at magnetic fields from 0.02 to 5 T, yielded magnetic moments of 5.976 and 5.974 Bohr magnetons for 1 and 2, respectively, clearly identifying them as high-spin (S = 5/2) ferric compounds. Variable-frequency (9.4 GHz and 34.5 GHz) EPR measurements, coupled with computer simulations, confirmed the magnetization results and yielded more precise values for the spin Hamiltonian parameters: gavg = 2.00 ± 0.03, |D| = 3.89 ± 0.09 cm−1, and E/D = 0.07 ± 0.01 for both compounds, where D and E are the axial and rhombic zero-field splittings. IR spectroelectrochemistry studies reveal that the first oxidations of these compounds occur at the porphyrin macrocycles and not at the Fe-NONOate moieties. Reactions of 1 and 2 with a histidine mimic (1-methylimidazole) generate RNO and NO, both of which may bind to the metal center if sterics allow, as shown by a comparative study with the Cupferron complex (T(p-OMe)PP)Fe(η2-ON(Ph)NO). Protonation of 1 and 2 yields N2O as a gaseous product, presumably from the initial generation of HNO that dimerizes to the observed N2O product.
Co-reporter:Prinson P. Samuel, Roman Neufeld, Kartik Chandra Mondal, Herbert W. Roesky, Regine Herbst-Irmer, Dietmar Stalke, Serhiy Demeshko, Franc Meyer, Vallyanga Chalil Rojisha, Susmita De, Pattiyil Parameswaran, A. Claudia Stückl, Wolfgang Kaim, Jonathan H. Christian, Jasleen K. Bindra and Naresh S. Dalal
Chemical Science (2010-Present) 2015 - vol. 6(Issue 5) pp:NaN3153-3153
Publication Date(Web):2015/03/20
DOI:10.1039/C5SC00646E
Cr(I)Cl is a very unstable species. The present work describes the stabilisation of Cr(I)Cl in the low coordinate environment of cyclic alkyl(amino) carbene ligands and its synthetic application to yield an unprecedented cationic complex with a two coordinate Cr(I). One electron reduction of (cAAC)2CrCl2 (1) with equivalent amount of KC8 results in the formation of (cAAC)2CrCl (2), with a distorted trigonal planar configuration at the metal centre. SQUID, EPR and theoretical studies reveal a Cr(I) centre with S = 5/2 spin ground state for 2. It represents the first example of a mononuclear Cr complex showing slow relaxation of magnetisation under an applied magnetic field. The chlorine atom in 2 is expected to be prone to further reactions with appropriate reagents. This qualifies 2 as a promising precursor for the preparation of various interesting complexes with Cr(I) in a low coordinate environment. The first example of this metathesis reaction is observed when 2 is treated with Na[B(C6H3(CF3)2)4] resulting in [(cAAC)2Cr]+[B(C6H3(CF3)2)4]−, a linear cationic complex with two coordinate Cr(I) and an S = 5/2 spin ground state.
.jpg)
![Nickel, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/2500/24803-99-4.png)
![Nickel, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/2500/24803-99-4_b.png)
![Zinc, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/1100000/17632-18-7.png)
![Zinc, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/1100000/17632-18-7_b.png)
![Pyridine, 2-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-](/data/chemimg/148200/1192206-01-1.png)
![Pyridine, 2-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-](/data/chemimg/148200/1192206-01-1_b.png)
![Pyridine, 2-[1-(phenylmethyl)-1H-1,2,3-triazol-4-yl]-](/data/chemimg/128700/862675-34-1.png)
![Pyridine, 2-[1-(phenylmethyl)-1H-1,2,3-triazol-4-yl]-](/data/chemimg/128700/862675-34-1_b.png)