Co-reporter:Nobuyuki Komine;Ayako Kuramoto;Kouhei Nakanishi;Masafumi Hirano
Topics in Catalysis 2014 Volume 57( Issue 10-13) pp:960-966
Publication Date(Web):2014 June
DOI:10.1007/s11244-014-0258-8
Smooth, reversible and Markovnikov selective alkene insertion of ethyl acrylate, acrylonitrile, and ethylene to MHCp(CO)3 (M = Mo, W) or MnH(CO)5 has been catalyzed by Pd(PPh3)4 at 20 °C. Cis-selective insertion of acetylene and dimethyl acetylenedicarboxylate into MHCp(CO)3 (M = Mo, W) also proceeded smoothly.
Co-reporter:Matthew T. Zamora, Kenta Oda, Nobuyuki Komine, Masafumi Hirano, Sanshiro Komiya
Journal of Organometallic Chemistry 2013 739() pp: 6-10
Publication Date(Web):
DOI:10.1016/j.jorganchem.2013.04.003
Co-reporter: Masafumi Hirano;Ryo Fujimoto;Kohei Hatagami;Nobuyuki Komine ; Sanshiro Komiya
ChemCatChem 2013 Volume 5( Issue 5) pp:1101-1115
Publication Date(Web):
DOI:10.1002/cctc.201200686
Abstract
A cationic complex [Ru{OC(O)CMeCH2-κ2O,O′}(PMe3)4]+CH2CMeCO2− (5 a) and its related carboxylato complexes are newly prepared by the reaction of [cis-RuH2(PMe3)4] (4) with carboxylic acids in methanol in 76–100 % yield. Complex 5 a reversibly transforms to the neutral form [cis-Ru{OC(O)CMeCH2-κ1O}2(PMe3)4] (2 a) in nonpolar solvents. Complex 2 a reversibly liberates a PMe3 group to give [Ru{OC(O)CMeCH2-κ1O}{OC(O)CMeCH2-κ2O,O′}(PMe3)3] (12 a) from which a stereoselective CH bond cleavage reaction occurs to give a ruthenalactone [Ru{OC(O)CMeCH-κ2O,C}(PMe3)4] (1 a) from the release of methacrylic acid. Complexes 2 a and 5 a also give 1 a but the prior dissociation of a PMe3 is indispensable for the CH bond cleavage reaction. Complex 1 a establishes an equilibrium with 2 a (or 5 a) in solution. In this reaction, one coordinated carboxylato ligand is engaged in the CH bond cleavage reaction as a proton acceptor, but neither the added carboxylato anion nor typical proton acceptors such as proton sponge assist the reaction. In [D4]MeOH, a catalytic stereospecific deuteration of carboxylic acids has been achieved by 4, in which the equilibrium between 5 a and 1 a plays a key role.
Co-reporter:Sanshiro Komiya
Coordination Chemistry Reviews 2012 Volume 256(5–8) pp:556-573
Publication Date(Web):March–April 2012
DOI:10.1016/j.ccr.2011.10.017
Synthesis, reactions, and catalyses of heterodinuclear organometallic complexes without connecting ligands L2RM-ML′n (M = Pt, Pd; M′ = Mo, W, Mn, Re, Fe, Co) are described. They are regarded as a simplest model for studying the cooperative effect of two transition metals in catalysis. Organic ligand R is found to move reversibly from M to M′ along M–M′ bond, which is accelerated by addition of electron-deficient olefins. The reaction is regarded as a reductive elimination of M′–R and the reverse process as oxidative addition reaction. The results may mimic the mobility of σ-bound organic group on heterogeneous surface. Visible light enhances reductive elimination (alkyl group transfer) in (tBu2bpy)Me2PhPt-Mn(CO)5. Enhanced CO insertion of heterodinuclear methylpalladium–cobalt system in comparison with mononuclear systems is presented and the mechanism is found to involve alkyl transfer followed by CO insertion into the Co–C bond and oxidative addition of σ-organic groups along heterometal–metal bond. Highly specific C–S bond cleavage reactions of heterodinuclear complex with thiiranes and thietanes are presented. Catalytic reactions such as carbonylation of thietane, copolymerization of aziridine and CO, and insertion of olefin or acetylene into Mo(or W)–H bond promoted by heterodinuclear complexes were described, where enhanced catalytic activity has been shown.Graphical abstract. Synthesis, reactions, and catalyses of heterodinuclear σ-bound organometallic complexes L2RM-ML′n (M = Pt, Pd; M′ = Mo, W, Mn, Re, Fe, Co), which have no connecting ligands for these metals, are described as one of simplest models for an active species in bimetallic catalysis, where reversible migration of σ-bound organic ligand R along M–M′ linkage, enhanced CO insertion, selective C–S bond cleavage of heterocycles, and a few catalytic reactions promoted by heterodinuclear complexes are described.Highlights► Heterodinuclear organometallic complexes without connecting ligands are synthesized. ► Reversible σ-bound organic ligand transfer from M to M′ is shown. ► Enhanced CO insertion and selective C–S bond cleavage of thiiranes are presented. ► Heterodinuclear complexes catalyze carbonylation of thietane. ► Pd-catalyzed insertion of olefin or acetylene into Mo(or W)–H bond is described.
Co-reporter:Masafumi Hirano, Masahiro Murakami, Toshinori Kuga, Nobuyuki Komine, and Sanshiro Komiya
Organometallics 2012 Volume 31(Issue 1) pp:381-393
Publication Date(Web):December 20, 2011
DOI:10.1021/om200974c
A series of RuCp[OC6H3(CH2CH═CH2-2)(R)](PPh3)n complexes (n = 2, R = H (1a); n = 1, R = 4-OMe (2b), 4-Me (2c), 4-Ph (2d), 4-Br (2e), 4-NO2 (2f), 6-OMe (2g), 6-Me (2h), 6-Ph (2i)) have been prepared in 27–76% yields. These 2-allylaryloxo complexes 1a and 2b–f are in equilibrium between RuCp[OC6H3(CH2CH═CH2-2)(R)-κ1O](PPh3)2 (1) and RuCp[OC6H3(CH2CH═CH2-2)(R)-κ1O,η2C,C′](PPh3) (2) in solution, and 2g–i do not react with PPh3. The equilibrium constant K1 (K1 = [2][PPh3]/[1]) is about the same for 1a and 2b–f (K1 = 0.07–0.31 M). In contrast to the conventional aryloxo complexes of the late transition metals, treatment of 1a and 2a–g with weak Brϕnsted acids (HOR) gives a rapid equilibrium with 2·HOR. The association constant K2 (K2 = [2·HOR]/([2][HOR])) increases on decreasing the pKa value of the acid employed and on increasing the induction effect of substituents at the 4-position in the aryloxo group. These features suggest present association being regarded as a simple acid–base interaction. Interestingly, further association of 2·HOR with the second acid leads to the cleavage of the benzylic C–H bond, giving RuCp[C3H4{1-C6H3(OH-2)(R)}-η3C,C′,C″](PPh3) (3). The thermodynamic and kinetic studies suggest formation of hydrogen bonds among two Brϕnsted acid molecules, lone-pair electrons in the aryloxo oxygen, and a benzylic methylene proton. Such association makes the Ru(II) center more electrophilic to attack the benzylic carbon to give 3.
Co-reporter:Nobuyuki Komine, Kohtaro Ishiguro, Sachiyo Kanai, Masafumi Hirano, Sanshiro Komiya
Journal of Organometallic Chemistry 2011 696(10) pp: 1927-1930
Publication Date(Web):
DOI:10.1016/j.jorganchem.2010.10.037
Co-reporter:Shin-ichi Tanaka, Nobuyuki Komine, Masafumi Hirano, Sanshiro Komiya
Journal of Organometallic Chemistry 2011 696(2) pp: 632-635
Publication Date(Web):
DOI:10.1016/j.jorganchem.2010.09.020
Co-reporter:Masafumi Hirano, Shin-ya Tatesawa, Minoru Yabukami, Yoko Ishihara, Yusuke Hara, Nobuyuki Komine, and Sanshiro Komiya
Organometallics 2011 Volume 30(Issue 19) pp:5110-5122
Publication Date(Web):September 12, 2011
DOI:10.1021/om200345h
The (2,6-dimethylbenzenethiolato)platinum(II) complexes PtR(SC6H3Me2-2,6-κ1S)L2 (R = Me, L = PMe3 (1a), PPh3 (1c), L2 = dppe (1d), dppp (1e); R = Et, L = PPh3 (2c); R = CH2CMe3, L = PPh3 (3c)) and Pt(SC6H3Me2-2,6-κ1S)2L2 (L = PMe3 (4a), PEt3 (4b), PPh3 (4c), L2 = dppe (4d), dppp (4e), dppb (4f)) have been prepared. Heating of these compounds results in an internal sp3 C–H bond cleavage reaction, giving the thiaplatinacycle complexes Pt[SC6H3(CH2-2)(Me-6)-κ2S,C]L2 (L = PMe3 (5a), PEt3 (5b), PPh3 (5c), L2 = dppe (5d), dppp (5e), dppb (5f)) in moderate to quantitative yields. The reactions of 1c and 4c proceed via prior dissociation of PPh3, and a concerted mechanism is proposed. Of particular interest is the sp3 C–H bond activation step, whose observed rate constant for 4c is no less than 104-fold faster than that for 1c. The arenethiolato group is expected to enhance the C–H bond cleavage step as a hydrogen acceptor.
Co-reporter:Masafumi Hirano, Sayaka Togashi, Muneaki Ito, Yuko Sakaguchi, Nobuyuki Komine and Sanshiro Komiya
Organometallics 2010 Volume 29(Issue 14) pp:3146-3159
Publication Date(Web):June 24, 2010
DOI:10.1021/om100249s
A series of bis(2,6-dimethylbenzenethiolato)ruthenium(II) complexes, Ru(SC6H3Me2-2,6-κ1S)2(TRIPHOS-κ3P,P′,P′′) (2a), Ru(SC6H3Me2-2,6-κ1S)2(TDPME-κ3P,P′,P′′) (2b), trans-Ru(SC6H3Me2-2,6-κ1S)2(DPPE-κ2P,P′)2 (3c) and Ru(SC6H3Me2-2,6-κ1S)2(PMe3)3 (2d) are prepared. Treatment of 2a with PMe3 in benzene at 50 °C results in the sp3 C−H bond cleavage reaction of the ortho methyl in thiolato group to give a stereochemical mixture of thiaruthenacycle complex Ru[SC6H3(CH2-2)(Me-6)-κ2S,C](TRIPHOS-κ3P,P′,P′′)(PMe3) (1a) in 70% yield ([(OC-6−34)-1a]/[(OC-6−25)-1a] = 80/20) with concomitant liberation of 2,6-dimethylbenzenethiol. Similar treatment of 2d with PMe3 in benzene at room temperature rapidly produced cis-Ru[SC6H3(CH2-2)(Me-6)-κ2S,C](PMe3)4 (1d) quantitatively. Treatment of 2b with PMe3 results in the formation of 1d by the C−H bond cleavage reaction and ligand displacement reaction. The C−H bond cleavage reaction does not occur from 3c under these conditions. Treatment of 2d with PMe3 in methanol does not give 1d at all but yields cis-Ru(SC6H3Me2-2,6-κ1S)2(PMe3)4 (3d), which is not responsible for formation of 1d, suggesting importance of coordinative unsaturation for the C−H bond cleavage reaction. The kinetic study suggests that present C−H bond cleavage reaction proceeds by a concerted mechanism.
Co-reporter:Masafumi Hirano, Izirwan Bin Izhab, Naoki Kurata, Kaori Koizumi, Nobuyuki Komine and Sanshiro Komiya
Dalton Transactions 2009 (Issue 17) pp:3270-3279
Publication Date(Web):14 Mar 2009
DOI:10.1039/B821179E
Insertion of a dimethyl acetylenedicarboxylate (DMAD) into the Ru–C bond in a cycloruthenated complex Ru[OC6H3(2-CH2)(6-Me)-κ2O,C](PMe3)4 (2) has been achieved to give a seven-membered oxaruthenacycle Ru[OC6H3{2-CH2C(CO2Me)C(CO2Me)}(6-Me)-κ2O,C](PMe3)3 (3) in 47% yield. The molecular structure of 3 by X-ray analysis shows an agostic interaction between the ruthenium and one of the benzylic methylene protons. Complex 3 shows fluxional behaviour in solution and the variable temperature NMR studies suggest this fluxionality to be responsible for the turnstile rotation of three PMe3 ligands and the rotation of the α-methoxycarbonyl group. Heating of a toluene solution of 3 at 100 °C for 2 h results in the 1,3-H shift reaction in 3 to give a κ1O,η3-C,C′,C″ allylic complex Ru[OC6H3{2-CHC(CO2Me)CH(CO2Me)}(6-Me)-κ1O,η3C,C′,C″](PMe3)3 (6) (80–90%), whose molecular structure is revealed by X-ray analysis. Acidolyses of 3 and 6 give 2-[(Z)-2′,3′-bis(methoxycarbonyl)allyl]-6-methylphenol (7) (88%) and 2-[(Z)-2′,3′-bis(methoxycarbonyl)propenyl]-6-methylphenol (8) (47%), respectively, and iodolyses of 3 and 6 produce 2,3-bis(methoxycarbonyl)-8-methyl-4H-benzopyran (9) (24%) and 2,3-bis(methoxycarbonyl)-8-methyl-2H-benzopyran (10) (48%), respectively.
Co-reporter:Sanshiro Komiya, Sei Ezumi, Nobuyuki Komine and Masafumi Hirano
Organometallics 2009 Volume 28(Issue 13) pp:3608-3610
Publication Date(Web):May 28, 2009
DOI:10.1021/om900319a
Novel visible light induced reductive elimination of a heterodinuclear triorganoplatinum−manganese complex, (tBu2bpy)Me2PhPt−Mn(CO)5, gives a methylmanganese complex, MnMe(CO)5, and a methylphenylplatinum complex, PtMePh(tBu2bpy).
Co-reporter:Masafumi Hirano, Yumiko Sakate, Nobuyuki Komine, Sanshiro Komiya and Martin A. Bennett
Organometallics 2009 Volume 28(Issue 17) pp:4902-4905
Publication Date(Web):August 4, 2009
DOI:10.1021/om9004065
The trans-bis(methoxycarbonyl)ruthenacyclopentane complex [trans-Ru{C2H{C(O1)OMe}C3H2C4H2C5H{C(O2)OMe}-κ1C2,κ1O1,κ1C5,μ-κ1O2}(η4-1,5-COD)]2 (2) and the acetonitrile adduct Ru[C2H(CO2Me)CH2CH2C5H(CO2Me)-κ1C2,κ1C5](η4-1,5-COD)(NCMe)2 (3) have been obtained from the reaction of Ru(η6-naphthalene)(η4-1,5-COD) (1) with methyl acrylate and are active for the Ru(0)-catalyzed dimerization of methyl acrylate. Thus, complex 3 catalyzes the tail-to-tail dimerization of methyl acrylate in 52% yield at 140 °C in THF, suggesting that this dimerization proceeds by the oxidative coupling mechanism.
Co-reporter:Shin-ichi Tanaka, Nobuyuki Komine, Masafumi Hirano and Sanshiro Komiya
Organometallics 2009 Volume 28(Issue 18) pp:5368-5381
Publication Date(Web):September 2, 2009
DOI:10.1021/om900421k
Heterodinuclear platinum (or palladium) complexes having a terminal carbene, (Ph3P)Cl(Me2NHC)M−M′L′n (M = Pt: M′L′n = Co(CO)4 (1), Mn(CO)5 (2), MoCp(CO)3 (3), FeCp(CO)2 (4); M = Pd: M′L′n = Co(CO)4 (33), Mn(CO)5 (34)) and (L)Cl{(PhCH2)YC}Pt−M′L′n (M′L′n = Co(CO)4, L = PMe2Ph: Y = NPh2 (5), OEt (6), OMe (7), OiPr (8); M′L′n = Co(CO)4, L = PPh3: Y = OEt (9); M′L′n = Mn(CO)5, L = PMe2Ph: Y = OEt (10)), are synthesized by metathesis reactions of dichlorocarbene complexes with metalate Na+[M′L′n]−. These complexes are characterized by NMR and IR spectroscopies and elemental analysis, and the molecular structures of 1, 2, and 6 are determined by X-ray structure analysis. Deprotonation of 6−10 with base gives heterodinuclear μ-alkoxystyrylplatinum−metal complexes, (L)(CO){μ-PhHC═YC}Pt−M′L′n−1 (M′L′n−1 = Co(CO)3, L = PMe2Ph: Y = OEt (13), OMe (14), OiPr (15); M′L′n−1 = Co(CO)3, L = PPh3: Y = OEt (16); M′L′n−1 = Mn(CO)4, L = PMe2Ph: Y = OEt (12)), where the E isomer is thermodynamically stable. Z isomers of corresponding monomeric alkoxystyrylplatinum(II) complexes are found to be more stable than the corresponding E isomers. In contrast, both the dinuclear diphenylaminostyrylplatinum−cobalt complex and its mononuclear analogue favor the Z configuration.
Co-reporter:Nobuyuki Komine ; Takuma Hirota ; Masafumi Hirano
Organometallics 2008 Volume 27(Issue 9) pp:2145-2148
Publication Date(Web):April 9, 2008
DOI:10.1021/om701062g
Oxidative addition of the Co−C bond of Co(η3-CH2CHCHR)(CO)3 (R = Ph (2a), Me (2b)) to Pt(styrene)(dppe) (1) gives (dppe)(μ-η1:η2-RCH═CHCH2)Pt−Co(CO)3 (R = Ph (3a), Me (3b)). The E-isomer of 3b showed fluxionality due to a site exchange process between the allyl carbon and Co at Pt and complete E-selective allyl transfer reaction from platinum to cobalt, which was accelerated by addition of acrylonitrile or PPh3, whereas the Z-isomer had a rigid structure and showed lower reactivity.
Co-reporter:Masafumi Hirano ; Toshinori Kuga ; Mariko Kitamura ; Susumu Kanaya ; Nobuyuki Komine
Organometallics 2008 Volume 27(Issue 15) pp:3635-3638
Publication Date(Web):July 10, 2008
DOI:10.1021/om800318z
Treatment of RuCp[OC6H4(CH2CH═CH2-2)-κ1O:η2C,C′](PPh3) (3c) with a Brønsted acid (HX) such as 2-allylphenol results in facile migration of a benzylic proton to the aryloxide, giving the (η3-allyl)ruthenium(II) complex RuCp[CH2CHCH(C6H4OH-2)-η3C,C′,C′′](PPh3) (4c). Thermodynamic and kinetic studies suggest that 3c associates with acid to give 3c·HX, and further addition of HX to 3c·HX causes the C−H bond cleavage reaction to give 4c.
Co-reporter:Nobuyuki Komine, Susumu Tsutsuminai, Hideko Hoh, Toshiyuki Yasuda, Masafumi Hirano, Sanshiro Komiya
Inorganica Chimica Acta 2006 Volume 359(Issue 11) pp:3699-3708
Publication Date(Web):1 August 2006
DOI:10.1016/j.ica.2005.12.026
Synthesis of a series of heterodinuclear phenylpalladium–molybdenum(or -tungsten) complexes having a bidentate nitrogen ligand, L2PhPd–MCp(CO)3 (M = Mo, L2 = tmeda (12), bpy (13), phen (14); M = W, L2 = tmeda (15)) by metathetical reactions of PdPhIL2 with Na[MCp(CO)3] and acylplatinum–molybdenum(or -tungsten) complex having a 1,2-bis(diphenylphosphino)ethane ligand, (dppe)(RCO)Pt–MCp(CO)3 (M = Mo, R = Et (16), CH2CMe3 (18); M = W, R = Et (17)) by CO insertion into Pt–C bond in corresponding alkyl analogues (dppe)RPt–MCp(CO)3 (M = Mo, R = Et (6), CH2CMe3, M = W, R = Et (8)) are described. These complexes are characterized by NMR and IR spectroscopies and elemental analyses, and the molecular structures of 12, 13, 17 and 18 are determined by X-ray structure analysis. The geometry at Pt (or Pd) is square planar and the MCp(CO)3 moiety has three-leg piano-stool geometries in these complex expect the propionylplatinum–tungsten complex 17, which shows apparent but unexpected structural deformation at Pt and W, giving twisted square-planar and four-leg piano-stool geometries for platinum and tungsten, respectively.Heterodinuclear phenypalladium–molybdenum(or -tungsten) or acylplatinum–molybdenum(or -tungsten) complexes, L2RM–M′Cp(CO)3 (M = Pd, Pt, M′ = Mo, W, L2 = tmeda, bpy, phen, dppe, R = Ph, COEt, COCH2Me3) have been synthesized by metathetical reactions of PdPhIL2 with Na[MCp(CO)3] or CO insertion into Pt–C bond in corresponding alkyl analogues. The propionylplatinum–tungsten complex, (dppe)(EtCO)Pt–WCp(CO)3 shows apparent but unexpected structural deformation at Pt and W, giving twisted square-planar and four-leg piano-stool geometries, respectively.
Co-reporter:Masaki Furuya, Susumu Tsutsuminai, Hiroto Nagasawa, Nobuyuki Komine, Masafumi Hirano and Sanshiro Komiya
Chemical Communications 2003 (Issue 16) pp:2046-2047
Publication Date(Web):09 Jul 2003
DOI:10.1039/B305799B
Heterodinuclear organoplatinum-cobalt complex having a 1,2-bis(diphenylphosphino)ethane ligand (dppe)MePt–Co(CO)4 catalyzes CO insertion into the C–S bond of thietanes in THF at 100 °C under 1.0 MPa of CO for 2 h to give γ-thiobutyrolactone in quantitative yield.
Co-reporter:Sanshiro Komiya and Masafumi Hirano
Dalton Transactions 2003 (Issue 8) pp:1439-1453
Publication Date(Web):11 Mar 2003
DOI:10.1039/B300601H
Ru(η4-1,5-cyclo-octadiene)(η6-1,3,5-cyclo-octatriene)
(1) is one of the most versatile zero-valent ruthenium complexes bearing two labile cyclopolyenes and acts as a potential precursor for catalytic processes involving bond cleavage reactions in the presence of suitable Lewis bases. However, detailed studies of the bond cleavage step had, until now, been relatively less explored at a molecular level. The present Perspective is an account of our recent studies concerning: (1) the reactions of 1 with Lewis bases, (2) carbon–oxygen, carbon–sulfur, oxygen–hydrogen, nitrogen–hydrogen and carbon–hydrogen bond cleavage reactions by 1 in the presence of tertiary phosphine, (3) selective sp3 carbon–hydrogen bond cleavage by 1 by use of co-ordination of an anchoring chalcogen atom, and (4) preparation of an enolatoruthenium(II) complex derived from 1 as an active intermediate in chemoselective catalytic Knöevenagel and Michael reactions.
Co-reporter:Masafumi Hirano, Koji Onuki, Yuichi Kimura, Sanshiro Komiya
Inorganica Chimica Acta 2003 Volume 352() pp:160-170
Publication Date(Web):6 August 2003
DOI:10.1016/S0020-1693(03)00132-4
Reactions of [Ru(1,5-COD)(1,3,5-COT)] (1) (COD=cyclooctadiene, COT=cyclooctatriene) with benzo[b]thiophene, thiophenes, benzo[b]furan, and furans in the presence of PEt3 result in the regioselective CH bond cleavage of these heterocyclic compounds giving [Ru(1-5-η5-C8H11)(R)(PEt3)2] [R=2-benzo[b]thienyl (4a), 2-thienyl (4b), 5-(2-ethoxylcarbonyl)thienyl (4c), 5-(2-acetyl)thienyl (4d), 5-(3-acetyl)thienyl (4e), 2-benzo[b]furyl (5a), 2-furyl (5b), 5-(2-acetyl)furyl (5c)]. Similar treatments of 1 with indoline and pyrrole lead to cleavage of the NH bond giving [Ru(1-5-η5-C8H11)(R)(PEt3)2] [R=1-indolyl (6a), 1-pyrrolyl (6b)]. The time-course study for the reaction of 1/PEt3 with benzo[b]thiophene monitored by use of NMR suggests prior formation of a zero-valent complex [Ru(1-4-η4-1,3,5-COT)(PEt3)3] (2a) followed by production of 4a. Kinetic study reveals that the rate of the reaction of 2a with benzo[b]thiophene is the first-order dependence on [2a], [benzo[b]thiophene], and [PEt3]−1, respectively. This fact suggests prerequisite dissociation of PEt3 from a coordinatively saturated complex 2a giving a vacant site for interaction of Ru center with benzo[b]thiophene.Regioselective C-H or N-H bond cleavage reactions of heterocyclic compounds by [Ru(1,5-COD)(1,3,5-COT]/monodentate phosphine give corresponding (h5-cyclooctadienyl) ruthenium(II) complexes Ru(1,5-cyclooctadienyl)(R)L2. Kinetic study reveals a mechanism involving prerequisite coordination of substrates for the bond cleavage reactions.9
Co-reporter:Masafumi Hirano, Sayori Kiyota, Masataka Imoto and Sanshiro Komiya
Chemical Communications 2000 (Issue 17) pp:1679-1680
Publication Date(Web):23 Aug 2000
DOI:10.1039/B004099L
Treatment of the N-bonded enolatoiron(II)
complex [M(H)(NCCHCO2R)(L)2] [1a, M = Fe, R = Me, L =
depe {1,2-bis(diethylphosphino)ethane}; 1b, M = Fe, R = Et, L =
depe; 2, M = Ru, L = dppe] with acrylonitrile results in mono-Michael
addition to give
[M(H){NCC(C2H4CN)CO2R}(L)2
], which is found to be an active intermediate for the catalytic
double-Michael reaction of cyanoacetate with acrylonitrile.
Co-reporter:Yoko Usui, Junko Noma, Masafumi Hirano, Sanshiro Komiya
Inorganica Chimica Acta 2000 Volume 309(1–2) pp:151-154
Publication Date(Web):20 November 2000
DOI:10.1016/S0020-1693(00)00248-6
Co-reporter:Masafumi Hirano, Naoki Kurata, Sanshiro Komiya
Journal of Organometallic Chemistry 2000 Volume 607(1–2) pp:18-26
Publication Date(Web):11 August 2000
DOI:10.1016/S0022-328X(00)00169-8
Successive OH and sp3 CH bond activation of ortho-substituted phenols has been achieved by the reactions of Ru(1,5-cyclooctadiene)(1,3,5-cyclooctatriene) (1) with 2,6-xylenol and 2-allylphenol in the presence of PMe3 giving oxaruthenacycle complexes such as cis-(H2)(6-Me)](PMe3)4 (4) or 3H4)](PMe3)3 (5), respectively. They are formed by the initial protonation of Ru(1-2-η2:5-6-η2-cycloocta-1,5-diene)(1-4-η4-cycloocta-1,3,5-triene)(PMe3) by phenols giving cationic (η5-cyclooctadienyl)ruthenium(II) complexes [Ru(η5-C8H11)(PMe3)3]+[OAr]−·(HOAr)n [Ar=C6H3Me2-2,6 (2a), C6H4(2-CH2CHCH2) (2b), C6H4{2-(E)-CHCHMe} (2c), Ph (2d); C6H4Me-2 (2e); C6H4(2-CHMe2) (2f), and C6H4(2-CMe3) (2g)] followed by sp3 CH bond cleavage reaction. The molecular structure of 2c reveals that the cyclooctadienyl group coordinates to the ruthenium center by an η5-fashion, where one equivalent of (E)-2-propenylphenol is associated with aryloxo anion. Further treatment of 2a and 2c with PMe3 results in the formation of oxaruthenacycle complexes to give 4 and 5, respectively. These facts clearly demonstrate that this sp3 CH bond cleavage reaction occurs at a divalent ruthenium center. On the other hand, reactions of 2d–g afford (hydrido)(aryloxo)ruthenium(II) complexes, cis-Ru(H)(OAr)(PMe3)4 [Ar=Ph (6a), C6H4Me-2 (6b), C6H4(2-CHMe2) (6c), C6H4(2-CMe3) (6d)].
Co-reporter:Takashi Morikita, Masafumi Hirano, Akito Sasaki, Sanshiro Komiya
Inorganica Chimica Acta 1999 Volume 291(1–2) pp:341-354
Publication Date(Web):August 1999
DOI:10.1016/S0020-1693(99)00146-2
Treatment of Fe(N2)(depe)2 [depe=1,2-bis(diethylphosphino)ethane] (1) with benzo[b]thiophene at room temperature results in the regioselective C–S and C–H bond cleavages giving H)(depe)2 (2a) and trans-FeH()(depe)2 (3a) in 72 and 19% yields, respectively. Complex 1 also reacts with thiophene, 2- and 3-acetylthiophenes and 2- and 3-methylthiophenes to give both C–S and C–H bond oxidative addition products: H)(depe)2 (2b) and trans-FeH()(depe)2 (3b), H](depe)2 (2c) and trans-FeH[](depe)2 (3c), H](depe)2 (2d) and trans-FeH[](depe)2 (3d), and H](depe)2 (2e) and trans-FeH[](depe)2 (3e), respectively. On the other hand, only C–H bond cleavage takes place in the reactions of 1 with furans such as furan, benzo[b]furan, and 2,3-dihydrofuran to give trans-FeH()(depe)2 (4a), FeH()(depe)2 (4b) and trans-FeH(H2)(depe)2 (4c) and N–H bond is exclusively cleaved by the reaction of 1 with pyrroles such as pyrrole, indole and 2-acetylpyrrole to give trans-FeH(H)(depe)2 (5a), trans- and cis-FeH(6H4)(depe)2 (5b) and FeH[H](η2-depe)(η1-depe) (6). Treatment of 2a with MeI results in the Fe–S bond cleavage of the thiaferracycle giving trans-FeI[(E)-CHCHC6H4-2-SMe](depe)2 (7) whose structure is unequivocally characterized by X-ray analysis. In contrast, hydrogenolysis of 2a with H2 (50 atm) leads to the cleavage of the Fe–C bond of the thiaferracycle to yield cis- and trans-FeH(SC6H4-2-Et)(depe)2 (8).
Co-reporter:Atsushi Fukuoka, Akihiro Sato, Kin-ya Kodama, Masafumi Hirano, Sanshiro Komiya
Inorganica Chimica Acta 1999 Volume 294(Issue 2) pp:266-274
Publication Date(Web):10 November 1999
DOI:10.1016/S0020-1693(99)00210-8
A series of organosiloxo complexes of platinum and palladium, MR(OSiPh3)(L2) (M=Pt, Pd; R=Me, Et, Ph; L2=cod, dppe), has been prepared and characterized. The square planar geometries of PtPh(OSiPh3)(cod) and PtEt(OSiPh3)(cod) are confirmed by X-ray structure analysis. In the reactions with hydrogen at 0°C and 1 atm, the siloxo complexes of Pt and Pd are reduced readily to give agglomerates of nanoclusters with complete hydrogenation of the ligands. The reduction activities of the siloxo and alkoxo complexes are higher than those of the corresponding alkyl complex PtMe2(cod). This high activity in reduction is applied to the preparation of supported Pt or Pd nanoclusters on silica, and the siloxo complexes adsorbed on silica are reacted with hydrogen at mild conditions. The resulting Pt/SiO2 gives a smaller mean diameter than that prepared from H2PtCl6/SiO2.
Co-reporter:Masafumi Hirano, Izirwan Bin Izhab, Naoki Kurata, Kaori Koizumi, Nobuyuki Komine and Sanshiro Komiya
Dalton Transactions 2009(Issue 17) pp:NaN3279-3279
Publication Date(Web):2009/03/14
DOI:10.1039/B821179E
Insertion of a dimethyl acetylenedicarboxylate (DMAD) into the Ru–C bond in a cycloruthenated complex Ru[OC6H3(2-CH2)(6-Me)-κ2O,C](PMe3)4 (2) has been achieved to give a seven-membered oxaruthenacycle Ru[OC6H3{2-CH2C(CO2Me)C(CO2Me)}(6-Me)-κ2O,C](PMe3)3 (3) in 47% yield. The molecular structure of 3 by X-ray analysis shows an agostic interaction between the ruthenium and one of the benzylic methylene protons. Complex 3 shows fluxional behaviour in solution and the variable temperature NMR studies suggest this fluxionality to be responsible for the turnstile rotation of three PMe3 ligands and the rotation of the α-methoxycarbonyl group. Heating of a toluene solution of 3 at 100 °C for 2 h results in the 1,3-H shift reaction in 3 to give a κ1O,η3-C,C′,C″ allylic complex Ru[OC6H3{2-CHC(CO2Me)CH(CO2Me)}(6-Me)-κ1O,η3C,C′,C″](PMe3)3 (6) (80–90%), whose molecular structure is revealed by X-ray analysis. Acidolyses of 3 and 6 give 2-[(Z)-2′,3′-bis(methoxycarbonyl)allyl]-6-methylphenol (7) (88%) and 2-[(Z)-2′,3′-bis(methoxycarbonyl)propenyl]-6-methylphenol (8) (47%), respectively, and iodolyses of 3 and 6 produce 2,3-bis(methoxycarbonyl)-8-methyl-4H-benzopyran (9) (24%) and 2,3-bis(methoxycarbonyl)-8-methyl-2H-benzopyran (10) (48%), respectively.

![Benzene, [(1E)-4,5-dimethyl-1,4-hexadienyl]-](http://img.cochemist.com/ccimg/135600/135519-74-3.png)
![Benzene, [(1E)-4,5-dimethyl-1,4-hexadienyl]-](http://img.cochemist.com/ccimg/135600/135519-74-3_b.png)



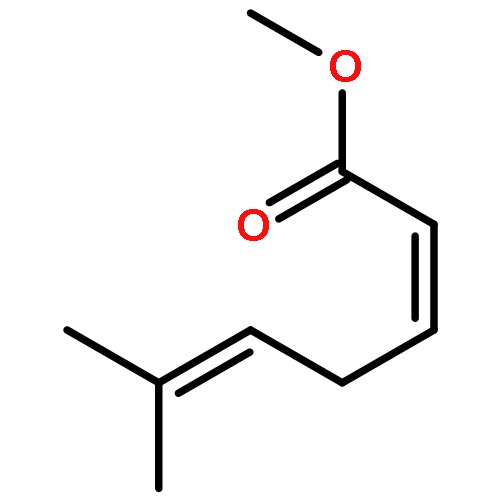

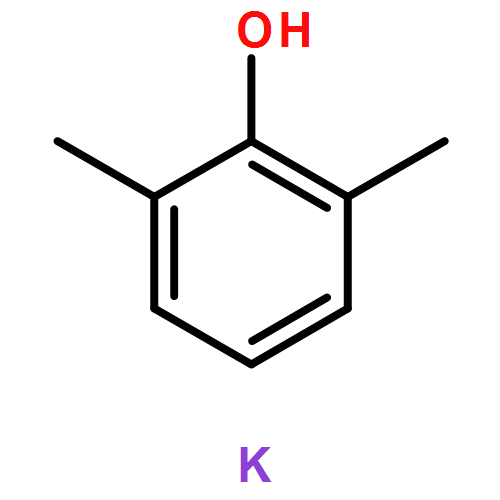
![Bicyclo[2.2.2]octane-2,5-dione](http://img.cochemist.com/ccimg/57400/57346-05-1.png)
![Bicyclo[2.2.2]octane-2,5-dione](http://img.cochemist.com/ccimg/57400/57346-05-1_b.png)
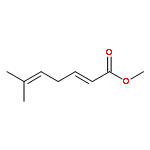

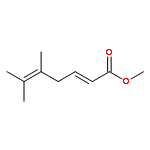












![bicyclo[2.2.2]octa-2,5-diene](http://img.cochemist.com/ccimg/600/500-23-2.png)
![bicyclo[2.2.2]octa-2,5-diene](http://img.cochemist.com/ccimg/600/500-23-2_b.png)
![Dibenzo[a,e]cyclooctene](http://img.cochemist.com/ccimg/300/262-89-5.png)
![Dibenzo[a,e]cyclooctene](http://img.cochemist.com/ccimg/300/262-89-5_b.png)