Co-reporter:Dongxia Ma;Zhe-Ning Chen;Xin Xu
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 3) pp:2417-2424
Publication Date(Web):2017/01/18
DOI:10.1039/C6CP06215F
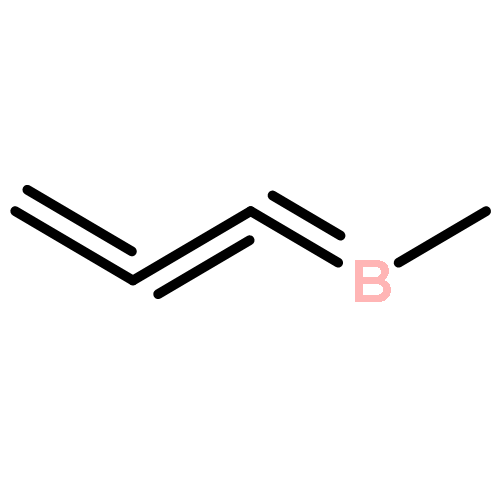
In this work, five new palladium(II) complexes have been designed as the model catalysts for methane to methyl trifluoroacetate conversion. All these compounds are analogues of the well-established (bis-NHC)PdBr2 complex (NHC, N-heterocyclic carbenes), derived by complexing the palladium(II) metal ion with the derivatives of bis-2-borabicyclo[1.1.0]but-1(3)-ene (bis-2BB) ligands using the sp2 carbons. Our density functional theory calculation results suggest that the (bis-2BB)PdBr2 catalysts outperform the popular (bis-NHC)PdBr2 complex in the desired catalytic process, and further reveal that the charge-shift bonding in the bis-2BB ligands contributes to the improved catalytic performance. These findings may spark new ideas for experimental design of more efficient organometallic catalysts for C–H bond activation and functionalization.
Co-reporter:Dongxia Ma, Congjie Zhang
Computational and Theoretical Chemistry 2017 Volume 1115(Volume 1115) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.comptc.2017.06.004
•The stable Pd(II)-catalysts bearing the bis-2-borabicyclo[1.1.0]but-1(3)-ene ligands were theoretically obtained.•The conversion of propane to propyl trifluoroacetate were investigated by density functional theory.•Pd(II)-complexes having the ligands with charge-shift bond are promising catalysts for the CH activation in propane.Using density functional theory (DFT), we have investigated the conversion of propane to propyl trifluoroacetate catalyzed by five kinds of (bis-2BB)PdBr2 complexes (bis-2BB is the derivatives of bis-2-borabicyclo[1.1.0]but-1(3)-ene). Calculated results indicate that the (bis-2BB)PdBr2 catalysts are superior to the popular (bis-NHC)PdBr2 complex in the desired catalytic process, consistent with the previous results of the conversion of methane to methyl trifluoroacetate catalyzed by (bis-2BB)PdBr2 complexes. Moreover, calculated natural charges and Wiberg bond index (WBI) values involving Pd(II) reveal that the charge-shift (CS) bonding characteristics in the bis-2BB ligands contributes to the improved catalytic performance of the (bis-2BB)PdBr2 complexes. Thus, we suggested that Pd(II)-catalysts having the ligands with CS bond might be promising organometallic catalysts for the activation and functionalization of CH bond in alkanes.Download high-res image (152KB)Download full-size image
Co-reporter:Congjie Zhang, Dongxia Ma
Journal of Organometallic Chemistry 2016 Volume 819() pp:242-247
Publication Date(Web):15 September 2016
DOI:10.1016/j.jorganchem.2016.07.011
•We successfully explained the BH activation in Ir(I) complexes.•Two kinds of methods to obtain B-σ-iridium complexes were given.•Dispersion correction is important for calculating the energy of iridium complexes.The structures, stability and activation of BH bonds of the twelve kinds of Ir(I) complexes supported by phosphine with o-carborane and carba-closo-dodecaborate ligand substituents, η2-C8H12Ir(I)Cl(PR2(CB11H11−)) and η2-C8H12Ir(I)Cl(PR2(C2B10H11)) (R = Me, iPr and CH(CF3)2) and η2-C8H12Ir(I)Cl(P(CH3)2X(CB11H11−)) and η2-C8H12Ir(I)Cl(P(CH3)2X(C2B10H11)) (X = CH2, NH and O) have been investigated using density functional theory (DFT) BP86 functional. Calculated results indicated that the BH bonds in η2-C8H12Ir(I)Cl(P(CH(CF3)2)2(CB11H11−)) and η2-C8H12Ir(I)Cl(PR2(C2B10H11)) (R = iPr and CH(CF3)2), as well as η2-C8H12Ir(I)Cl(P(CH3)2R(CB11H11−)) and η2-C8H12Ir(I)Cl(P(CH3)2R(C2B10H11)) (R = CH2, NH and O) are both thermodynamically and dynamically favorable to be activated, in which the oxidation addition of BH bond in η2-C8H12Ir(I)Cl(P(iPr)2(C2B10H11)) is consistent with the experimental results. In contrast, the BH bonds in η2-C8H12Ir(I)Cl(P(R)2(CB11H11−)) (R = Me and iPr) and η2-C8H12Ir(I)Cl(P(CH3)2(C2B10H11)) cannot be activated, in which η2-C8H12Ir(I)Cl(P(iPr)2(CB11H11−)) is consistent with the experimental result. The activation of BH bonds in carba-closo-dodecaborate phosphine and o-carborane phosphine of Ir(I) complexes depend up the distance between BH bond and Ir(I). Moreover, two kinds of approaches to obtain B-σ-irridium (III) complexes were given by the activation of BH bonds in Ir(I) complexes.
Co-reporter:Xiaofang Jia, Congjie Zhang
Computational and Theoretical Chemistry 2016 Volume 1075() pp:47-53
Publication Date(Web):1 January 2016
DOI:10.1016/j.comptc.2015.11.010
•The bonding of TML2 and TML2+ with planar tetracoordinate carbons (TM = Cu, Ag and Au) was studied.•The first electron transition energies of TML2+ associated with d–π transition are close to 260 nm.•The stable Au(III) complexes with planar tetracoordinate carbons were obtained.The structures and bonding of the complexes TML2 and TML2+ with planar tetracoordinate carbons (TM = Cu, Ag and Au; L = C3H2BCH3 and C3Me2BCMe3) have been investigated using density functional theory M06 method, in which L are the derivatives of 2-borabicyclo[1.1.0]but-1(3)-ene. The lengths of Au–C and Ag–C bonds in AuL2 and AgL2 are longer than those of in TML2+, whereas the lengths of Cu–C bonds in TML2 are shorter than those of in TML2+. The NBO analyses of TML2 and TML2+ reveal that the Cu–C bonds in CuL2 are conjugated π bonds, whereas Ag–C and Au–C in TML2 (TM = Ag and Au) are σ bonds. The first electron transition energies of TML2+ result from the d–π transition, and the wavelengths are close to 260 nm. Theoretically, the variations of the Gibbs free energies of the reactions of Ag(C3H2BCH3)2+ and Ag(C3Me2BCMe3)2+ with (tht)AuBr indicate that the two reactions are thermodynamically favorable. We also theoretically obtained the stable Au(III) complexes (C3H2BCH3)2AuBr2+ and (C3Me2BCMe3)2AuBr2+ with planar tetracoordinate carbons from the reactions of Au(C3H2BCH3)2+ and Au(C3Me2BCMe3)2+ with CsBr3.
Co-reporter:Shaoni Yang and Congjie Zhang
The Journal of Physical Chemistry A 2015 Volume 119(Issue 33) pp:8950-8957
Publication Date(Web):July 28, 2015
DOI:10.1021/acs.jpca.5b06449
Ten derivatives of 2-borabicyclo[1.1.0]but-1(3)-ene (1a–1j) with different degrees of frustration have been investigated using density functional theory. Moreover, 1a–1j as Lewis bases were used to form Lewis adducts C3X2BYH/B(C6F5)3 (2a–2j) with Lewis acid B(C6F5)3. Optimized geometries and the thermodynamic properties of giving the Lewis adducts C3X2BYH/B(C6F5)3 reveal that 2a–2j are frustrated Lewis pairs (FLPs). Their reactivity of activating H2 and HF show that 2a–2j are unfavorable to heterolytically cleave H2, whereas 2c–2j can cleave HF to form [C3X2BYH]+[FB(C6F5)3]−. In addition, we found the structures of [C3X2BYH]+ in [C3X2BYH]+[FB(C6F5)3]− contained a planar tetracoordinate carbon (ptC). Therefore, a new method of obtaining main group element compounds with ptC by using FLPs was presented.
Co-reporter:Congjie Zhang, Lichao Ning, and Jinxia Li
Organometallics 2013 Volume 32(Issue 23) pp:7083-7089
Publication Date(Web):November 14, 2013
DOI:10.1021/om400807w
Using density functional theory, the structures and stability of [1.1.1]propellanes C5H6 and E5Me6 (E = Si, Ge, Sn), as well as their dianions have been studied. Calculated results showed that the energy of Sn5Me62– is lower than that of Sn5Me6. In contrast, C5H62– and E5Me62– (E = Si, Ge) are electronically unstable, which successfully explains the experimental results of the reactions of E5R6 (E = Ge, Sn) with [FeCp(CO)2]2 and [RuCp(CO)2]2. Furthermore, FeCp(CO)2 and CH3 groups were introduced to capture the covalent resonance structures of C5H6 and E5Me6. Additionally, the ionic resonance structures of C5H6 and E5Me6 were captured by the strongly electron donating group (HCNMe)2C: and electron accepting group (AgCl) simultaneously. Hence, the “inverted” C–C and E–E bonds in [1.1.1]propellanes C5H6 and E5Me6 are covalent–ionic mixes. Together with their electron density and Laplacian values, direct evidence for the charge-shift bonding of the inverted C–C (C5H6) and E–E bonds (E5Me6) was presented.
Co-reporter:Congjie Zhang and Feifei Li
The Journal of Physical Chemistry A 2012 Volume 116(Issue 36) pp:9123-9130
Publication Date(Web):August 22, 2012
DOI:10.1021/jp305871m
We have designed a family of novel molecules that can act as a donor at a carbon atom to coordinate with Ag(I) and Au(I) to achieve stable complexes with a planar tetracoordinate carbon (ptC). Then, a kind of new strategy for the design of the compound with a ptC was provided.
Co-reporter:Pei Wang, Congjie Zhang
Journal of Molecular Structure: THEOCHEM 2010 Volume 955(1–3) pp:84-90
Publication Date(Web):15 September 2010
DOI:10.1016/j.theochem.2010.06.006
The B- and N-doped (4,4)i and (8,0)i (i = 3–5) carbon nanotubes, BCyN (y = 1–4, 6 and 8), B3CyN3 (y = 2 and 4), B2CN2, as well as BN nanotubes have been investigated using B3LYP/6-31G∗ method. The lowest energy structures of BxCyNx nanotubes of doped (8,0)i carbon nanotubes indicate that the B and N atoms prefer to dope (8,0)i carbon nanotubes from bottom to top layer by layer. When the number of BN layers of doping (4,4)i carbon nanotube is equal to i − 1, the B and N atoms are situated in the middle positions of BxCyNx nanotube and carbon atoms locate on its top and bottom. Otherwise, the doped ways of B- and N-doped (4,4)i carbon nanotubes are similar to those of (8,0)i. Calculated energy gaps of (4,4)i and (8,0)i (i = 3–5) BN nanotubes are between 5.51 and 6.38 eV, which are in good agreement with the experimental energy gap of 5.5 eV. In addition, the energy gaps of BC2N and BCN nanotubes are consistent with the previous theoretical and experimental results. The variety of energy gaps of BxCyNx nanotubes of doped (4,4)i and (8,0)i (i = 3–5) carbon nanotubes follows the same trend. However, the energy gaps of doped (4,4)i carbon nanotubes are greater than those of doped (8,0)i ones.
Co-reporter:Congjie Zhang, Pei Wang, Jinxia Liang, Wenhong Jia, Zexing Cao
Journal of Molecular Structure: THEOCHEM 2010 Volume 941(1–3) pp:41-46
Publication Date(Web):15 February 2010
DOI:10.1016/j.theochem.2009.10.036
Based on the most stable structure of C3B2H4 and its substitution of H with -Cl, -CH2OH, -CHOH, -CO and -COOH groups, a series of derivatives containing the planar tetracoordinate carbon (ptC) atoms as well as crown ether-like compounds from the assembling of C3B2H4 units have been constructed. At the B3LYP/6-311++G** and MP2/6-31G** levels of theory, these ptC compounds were predicted to be stable and they generally have large HOMO–LUMO gaps. The IR characteristic bands arising from the symmetrical and asymmetrical stretching vibrations of C–ptC, the stretching vibrations of C–B and B–H as well as the breath vibration of the two three-membered rings of C3B2 appear at 1000, 1250, 1600, 2800, and 1700 cm−1, respectively. Calculations also show that these ptC molecules have strong aromaticities and the ptC atom obeys the octal rule. Furthermore, the derivatives C3B2H2(COOH)2 and tetra-C3B2H2-16-crown-4 can serve as the chelate ligands and form stable complexes with uranyl.
Co-reporter:Congjie Zhang, Wenhong Jia and Zexing Cao
The Journal of Physical Chemistry A 2010 Volume 114(Issue 30) pp:7960-7966
Publication Date(Web):July 9, 2010
DOI:10.1021/jp102678v
We have investigated the structures, stabilities, aromaticities, and Wiberg bond indices of four types of compounds (8-like, trapezia, umbrella-like, and quadrangle) containing planar tetracoordinate carbon (ptC), and the stability rules for the compound with ptC were concluded on the basis of extensive calculations. Generally, the stability or viability of compound with ptC strongly depends on the number of three-membered ring and conjugated three-membered ring, as well as π electrons. These rules can be successfully used to identify the stability of other compounds reported in previous studies. On the basis of these rules, eight stable compounds with planar tetracoordinate nitrogen (ptN) are successfully constructed.
Co-reporter:Congjie ZHANG;Wenhong JIA ;Zexing CAO
Chinese Journal of Chemistry 2009 Volume 27( Issue 5) pp:882-886
Publication Date(Web):
DOI:10.1002/cjoc.200990148
Abstract
Fullerene[51] containing quasi-planar tetracoordinate carbons and its eight modificational compounds with quasi-planar tetracoordinate carbons have been constructed. Using B3LYP method, their structures, stabilities, and bonding properties have been investigated, indicating that the nine novel structures are stable and have big vertical detachment energies and relatively small vertical electron affinities. The structures of the nine compounds contain five- and six-membered rings, as well as bigger ten-membered rings.
Co-reporter:Jinxia Li, Congjie Zhang
Computational and Theoretical Chemistry (15 February 2014) Volume 1030() pp:
Publication Date(Web):15 February 2014
DOI:10.1016/j.comptc.2013.12.020
•The stable structures of (BGe3H6N)n (n = 1–4) and their electron spectra were obtained.•The stable structures and electron spectra of H3N(BGe3H6N)nBH3 and OC(BGe3H6N)nAgCl (n = 1–4) were obtained.•The one-dimensional periodic structures of [(BGe3X6N)2]n (X = H, F, Cl, Br and I) might be direct-gap semiconductor.The structures, stability and electronic spectra of the lantern-like molecule BGe3H6N [1.1.1]propellane, and its polymers (BGe3H6N)n, H3N(BGe3H6N)nBH3 and OC(BGe3H6N)nAgCl (n = 1, 2, 3 and 4) have been investigated using density functional theory B3LYP method. Calculated results indicate that (BGe3H6N)n, H3N(BGe3H6N)nBH3 and OC(BGe3H6N)nAgCl (n = 1, 2, 3 and 4) are situated at the minima on the potential energy surfaces. With the increase of n, the vertical transition energies decrease from 264.2 to 4787.4 nm for (BGe3H6N)n, 391.7 to 619.4 nm for H3N(BGe3H6N)nBH3 and 432.3 to 2850.4 nm for OC(BGe3H6N)nAgCl, respectively. In addition, energy bands of [(BGe3X6N)2]n (X = H, F, Cl, Br and I) with one-dimensional periodic structures show that the direct-gap of [(BGe3X6N)2]n is the same to its indirect-gaps, which the band gaps are 2.67, 2.94, 2.80, 2.37 and 1.36 eV, respectively. Therefore, [(BGe3X6N)2]n (X = H, F, Cl, Br and I) with one-dimensional periodic structures might be promising direct-gap semiconductor materials.Graphical abstract
Co-reporter:Congjie Zhang, Dongxia Ma, Shaoni Yang, and Jinxia Liang
ACS Omega Volume 1(Issue 4) pp:620-625
Publication Date(Web):October 19, 2016
DOI:10.1021/acsomega.6b00170
We have theoretically investigated the stability, chemical bonding, and coordination ability of the 2-Me-2-borabicyclo[1.1.0]but-1(3)-ene (2-Me-2BB) molecule using density functional theory and ab initio molecular dynamics (AIMD) simulations. Calculated results indicated that 2-Me-2BB is both thermodynamically and kinetically stable. The C═C bonds in 2-Me-2BB contain a π bond and a charge shift (CS) bond, different from those in 1-Me-borirene and cyclopropylene. Moreover, 2-Me-2BB can be a σ donor, leading to the formation of TM(2-Me-2BB)Ln complexes containing planar tetracoordinate carbon (ptC) with transition metals (TM = Sc–Cu), in which the lone electron pair of 2-Me-2BB results from its ionic resonance form. The lengths and Wiberg bond indices of the TM-ptC bond in TM(2-Me-2BB)Ln (TM = Sc–Cu) reveal that 2-Me-2BB can be a ligand similar to N-heterocyclic carbene. Therefore, 2-Me-2BB and its derivatives are promising molecules to obtain complexes with ptC. The natural charges on TM atoms in TM(2-Me-2BB)Ln (TM = Sc–Cu) complexes range from −0.97 to 1.54e, indicating that such complexes with ptC might have potential applications in catalytic chemistry.Topics: Free energy; Molecular structure; Potential energy; Reaction mechanism; Transition metal complexes (Inorg.); Vibrational frequency;
Co-reporter:Dongxia Ma, Congjie Zhang, Zhe-Ning Chen and Xin Xu
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 3) pp:NaN2424-2424
Publication Date(Web):2016/12/08
DOI:10.1039/C6CP06215F
In this work, five new palladium(II) complexes have been designed as the model catalysts for methane to methyl trifluoroacetate conversion. All these compounds are analogues of the well-established (bis-NHC)PdBr2 complex (NHC, N-heterocyclic carbenes), derived by complexing the palladium(II) metal ion with the derivatives of bis-2-borabicyclo[1.1.0]but-1(3)-ene (bis-2BB) ligands using the sp2 carbons. Our density functional theory calculation results suggest that the (bis-2BB)PdBr2 catalysts outperform the popular (bis-NHC)PdBr2 complex in the desired catalytic process, and further reveal that the charge-shift bonding in the bis-2BB ligands contributes to the improved catalytic performance. These findings may spark new ideas for experimental design of more efficient organometallic catalysts for C–H bond activation and functionalization.